
Unit-II: Elimination, Bioavailability and Bioequivalence (10 Hrs.)
Syllabus:
Elimination: Drug metabolism and basic understanding metabolic pathways, renal excretion of drugs, factors affecting renal excretion of drugs, renal clearance, Non renal routes of drug excretion of drugs.
Bioavailability and Bioequivalence: Definition and Objectives of bioavailability, absolute and relative bioavailability, measurement of bioavailability, in-vitro drug dissolution models, in-vitro–in-vivo correlations, bioequivalence studies, methods to enhance the dissolution rates and bioavailability of poorly soluble drugs.
Biotransformation of Drugs/ Metabolism
- The onset of pharmacological response depends upon two pharmacokinetic processes—
- Drug absorption, and
- Drug distribution (since most sites of action are in the extravascular tissues).
- The duration and intensity of action depend upon –
- Tissue redistribution of drug, and
- The rate of drug removal from the body/site of action, i.e. rate of elimination.
- Elimination is the major process for removal of a drug from the body and termination of its action. It is defined as the irreversible loss of drug from the body. Elimination occurs by two processes namely biotransformation and excretion.
- Biotransformation of drugs is defined as the chemical conversion of one form to another. The term is used synonymously with metabolism.
- The chemical changes are usually affected enzymatically in the body and thus, the definition excludes chemical instability of a drug within the body; for e.g. conversion of penicillin to penicilloic acid by the bacterial penicillinase and mammalian enzymes is metabolism but its degradation by the stomach acid to penicillenic acid is chemical instability.
Need for Drug Biotransformation
- All chemical substances that are not nutrients for the body and enter the body through, ingestion, inhalation or absorption are called as xenobiotics(Greek: xenos = foreign) orexogenous compounds.
- Drugs are also xenobiotics which enter the body by virtue of theirlipophilicity. It is interesting to note that for effective absorption, a drug needs to be sufficiently lipid soluble, but it is this same physicochemical property that enables it to bypass excretion.
- This is because only water-soluble agents undergo renal excretion (major route for exit of drugs from the body) whereas lipid soluble substances are passively reabsorbed from the renal tubules into the blood after glomerular filtration. Thus, if such a phenomenon continues, drugs would accumulate in the body and precipitate toxic reactions.
- However, to prevent such a consequence, the body is armed with the metabolic system which transforms the water insoluble, lipophilic, nonpolar drugs into polar and water-soluble products that can be easily excreted by the kidneys and are poorly reabsorbed; for instance, hippuric acid, the metabolite of benzoic acid, is 2.5 times more water-soluble. Drug biotransformation is thus a detoxification process
- Disposition of drug in the body as a consequence of metabolism is illustrates in the Fig. 1.
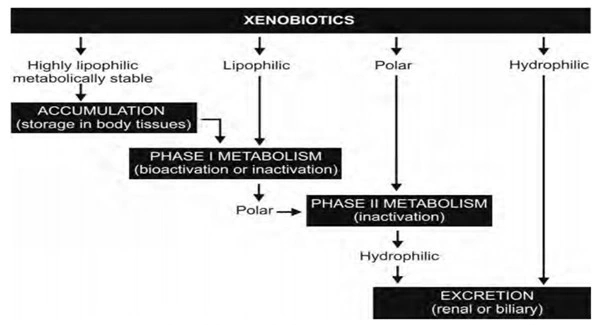
Fig. 1.Disposition of drug in the body as a consequence of metabolism
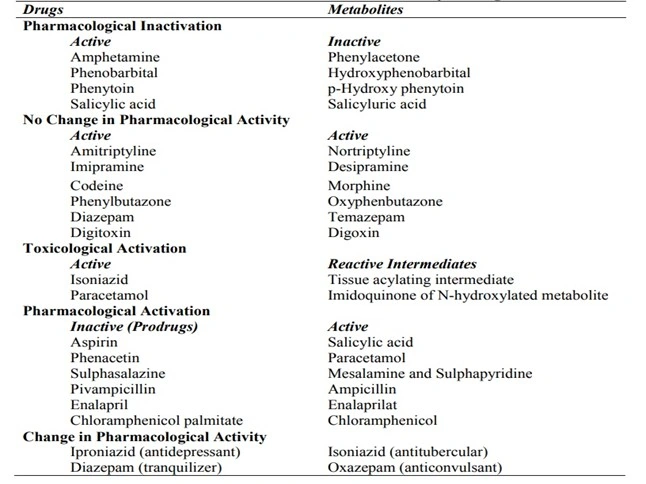
- Biotransformation –
- Normally results in pharmacological inactivation of drugs, i.e. it results in formation of metabolites with little or no pharmacological activity; e.g. conversion of phenytoin to p-hydroxy phenytoin.
- Occasionally yields metabolites with equal activity; e.g. conversion of phenylbutazoneto oxyphenbutazone.
- Rarely leads to toxicological activation of drugs, i.e. it results in formation of metabolites with high tissue reactivity; e.g. conversion of paracetamol to reactive metabolites that cause hepatic necrosis.
- Inactive drugs (prodrugs) also depend upon biotransformation for activation, the process being called as pharmacological activation; A change in pharmacological activity of the drug on metabolism has also been observed.
- TABLE 1. Metabolites and Relative Activity of Drugs
Drug Metabolising Organs
- Liver is the primary site for metabolism of almost all drugs (and other xenobiotics) because of its relative richness in possessing a large variety of enzymes in large amounts. Metabolism by organs other than liver (called as extrahepatic metabolism) is of minor importance sincelower level of drug metabolising enzymes are present in such tissues. The decreasing order of drug metabolising ability of various organs is:
- Liver > Lungs > Kidneys > Intestine > Placenta > Adrenals > Skin. Brain, testes, muscles, spleen, etc. also metabolise drugs but to a small extent.
Drug Metabolising Enzymes
- The enzymes that biotransform xenobiotics differ from those that metabolise food materials. They are versatile and non-specific in metabolising a large number of drugs. The enzymes are broadly divided into 2 categories:
- Microsomal enzymes (within the smooth endoplasmic reticulum)
- Non-microsomal enzymes. (outside the smooth endoplasmic reticulum)
CHEMICAL PATHWAYS OF DRUG BIOTRANSFORMATION
- R.T.Williams, the leading pioneer in drug biotransformation research, divided the pathways ofdrug metabolism reactions into two general categories—
- Phase I reactions, and
- Phase II reactions.
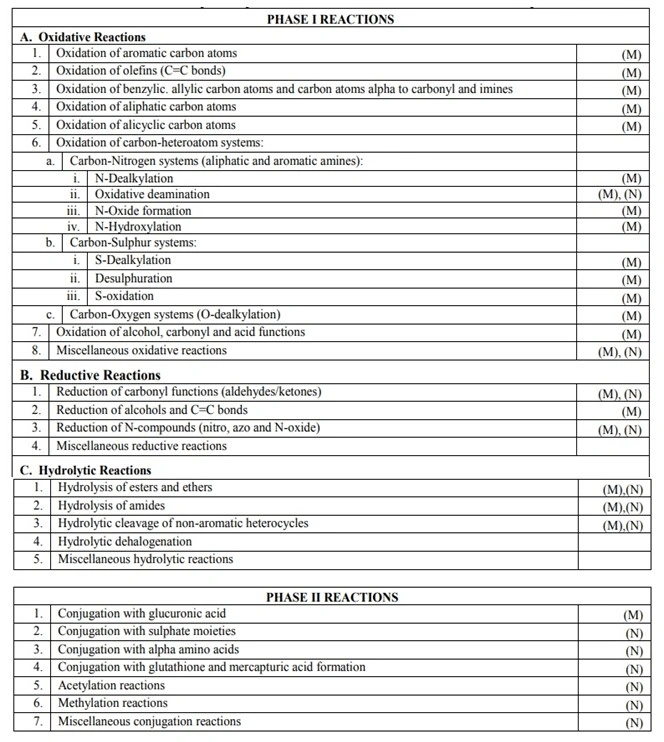
Phase I Reactions
- These reactions generally precede phase II reactions and include oxidative, reductive and hydrolytic reactions. By way of these reactions, a polar functional group is either introduced or unmasked if already present on the otherwise lipid soluble substrate, e.g. -OH (hydroxyl), -COOH (carboxyl), -NH2 (amino) and -SH (sulfhydryl). Thus, phase I reactions are also called as functionalization reactions.
- These transformations are also called as asynthetic reactions, opposite to the synthetic phase II reactions. The resulting product of phase I reaction is susceptible to phase II reactions.
Phase II Reactions
- These reactions generally involve covalent attachment of small polar endogenous molecules such as glucuronic acid, sulphate, glycine, etc. to either unchanged drugs or phase I products having suitable functional groups viz. -OH, -COOH, -NH2 and -SH and form highly water-soluble conjugates which are readily excretable by the kidneys (or bile). Thus, these reactions are called as conjugation reactions.
- Since the outcome of such processes are generally products with increased molecular size, they are also called as synthetic reactions. Quite often, a phase I reaction may not yield a metabolite that is sufficiently hydrophilic or pharmacologically inert but conjugation reactions generally result in products with total loss of pharmacological activity and high polarity. Hence, phase II reactions are better known as true detoxification reactions.
- Since these reactions generally involve transfer of moieties to the substrate to be conjugated, the enzymes responsible are called as transferases.
- The various phase I and phase II reactions are listed in Table 2
TABLE 2. Chemical Pathways of Drug Biotransformation — (M) and (N) Indicate Reactions Catalysed by Microsomal and Non-microsomal Enzymes.
PHASE I REACTIONS
Oxidative Reactions
- Oxidative reactions are the most important and most common metabolic reactions. Almost all drugs that undergo phase I biotransformation undergo oxidation at some stage or the other. A simple reason for oxidation being a predominant reaction is that energy in animals is primarily derived by oxidative combustion of organic molecules containing carbon and hydrogen atoms.
- Oxidative reactions increase hydrophilicity of xenobiotics by introducing polar functional groups such as—OH. Such a polar metabolite can thus rapidly undergo phase II reaction or is excretable by the kidneys.
- Oxidation of xenobiotics is non-specifically catalysed by a number of enzymes located in the microsomes. Such enzymes require both molecular oxygen (O2) and the reducing agent NADPH (Nicotinamide Adenine Dinucleotide Phosphate) to effect reaction. They are therefore referred to as the mixed-function oxidases. The overall stoichiometry of this reaction involving the substrate RH which yields the product ROH, is given by the following equation:
RH + O2 + NADPH + H+ → ROH + H2O + NADP+
Reductive Reactions
- Reductive metabolic reaction can generate polar functional groups such as hydroxy and amino. These groups can undergo further phase I or phase II conjugation reactions. Reduction reaction occurs on various functional groups like aldehyde, ketone, hydroxyl and nitro groups.
Hydrolytic Reactions
- These reactions differ from oxidative and reductive reactions in 3 respects:
- The reaction does not involve change in the state of oxidation of the substrate.
- The reaction results in a large chemical change in the substrate brought about by loss of relatively large fragments of the molecule.
- The hydrolytic enzymes are also involved in metabolism of endogenous substrates and these are found in liver, kidney and intestine.
- A number of functional groups are hydrolysed namely- esters, ethers, amides, hydrazides, etc.
PHASE II REACTIONS
- Phase II reactions involve transfer of a suitable endogenous moiety such as glucuronic acid, sulphate, glycine, etc. in presence of enzyme transferase to drugs or metabolites of phase I reactions having suitable functional groups to form highly polar, readily excretable and pharmacologically inert conjugates.
- Phase II reactions are the real drug detoxication pathways because–
- The conjugates/products of phase II reactions are absolutely free of pharmacological activity.
- The conjugates/products of phase II reactions are highly polar and thus easily excretable either in bile or urine.
- Tissue-reactive and carcinogenic metabolites formed as a result of phase I reaction are rendered harmless by conjugation with moieties such as glutathione.
- The order of capacities of important conjugation reactions is –
Glucuronidation > Amino Acid Conjugation > Sulphation and Glutathione Conjugation
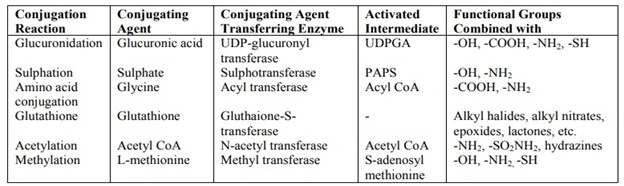
- Table 3 compares the various phase II reactions.
- The molecular weight of the conjugate is important in dictating its route of excretion –
- High molecular weight conjugates (>350) are excreted predominantly in bile
- Low molecular weight conjugates (<250) are excreted in urine.
- Thus, glutathione conjugates are always excreted in bile.
Table 3-Phase II Reactions and their Characteristics
Conjugation With Glucuronic Acid
- Also called as glucuronidation, it is the most common and most important phase II reaction for several reasons:
- Readily available source of conjugating moiety, D-glucuronic acid which is derived from D-glucose.
- Several functional groups viz. alcohols, acids, amines, etc. can combine easily with D-glucuronic acid.
- Quantitatively, conjugation with D-glucuronic acid occurs to a high degree.
- All mammals have the common ability to produce glucuronides,
- The free carboxyl function of glucuronic acid has a pKa in the range 3.5 to 4.0 and hence ionisable at both plasma and urine pH thereby greatly increasing the water solubility of the conjugated substrate.
- The glucuronidation enzymes are in close association with the microsomal mixed function oxidases, the major phase I drug metabolising enzyme system; thus, a rapid conjugation of phase I metabolites is possible.
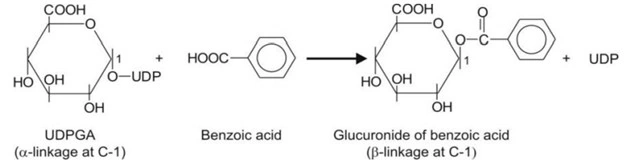
- Lastly, glucuronidation can take place in most body tissues since the glucuronic acid donor, UDPGA is produced in processes related to glycogen synthesis and thus, will never be deficient unlike those involved in other phase II reactions.
- Glucuronide formation occurs in 2 steps –
- Synthesis of an activated coenzyme uridine-5′-diphospho-α -D-glucuronic acid(UDPGA) from UDP-glucose (UDPG). The coenzyme UDPGA acts as the donor of glucuronic acid. UDPG is synthesized by interaction of α-D-glucose-1-phosphate with uridine triphosphate (UTP).
- Transfer of the glucuronyl moiety from UDPGA to the substrate RXH in presence of enzyme UDP-glucuronyl transferase to form the conjugate. In this step, the – configuration of glucuronic acid undergoes inversion and thus, the resulting product is – β-D-glucuronide (also called as glucosiduronic acid or glucopyranosiduronic acid conjugate).
The steps involved in glucuronide synthesis are depicted below:

where X = O, COO, NH or S.
An example of glucuronidation of benzoic acid is shown below.
Conjugation With Sulphate Moieties
- Sulphation is similar to glucuronidation but it is catalysed by nonmicrosomal enzymes andoccurs less commonly as the moiety that transfers sulphate to the substrate is easily depleted. This process is thus, easily saturable in comparison to glucuronidation.
- Sulphation is dominant at low substrate concentration, whereas
- Glucuronidation is dominant at high substrate concentration.
- Like glucuronidation, sulphation also occurs in 2 steps:
- Synthesis of an activated coenzyme 3′-phosphoadenosine-5′-phosphosulphate (PAPS)which acts as a donor of sulphate to the substrate.
- Transfer of sulphate group from PAPS to the substrate RXH in presence of enzymesulphotransferase (sulphokinase) and subsequent liberation of 3′-phosphoadenosine-5′-phosphate (PAP).
The steps are summarized in the equations below:

where X = O, NH
Conjugation With Alpha Amino Acids
- This reaction also occurs to a limited extent because of limited availability of amino acids.
- The reaction occurs in two steps:
- Activation of carboxylic acid drug substrate with ATP and coenzyme A (CoA) to forman acyl CoA intermediate. Thus, the reaction is a contrast of glucuronidation and sulphation where the donor coenzyme is activated and not the substrate.
- Acylation of the -amino acid by the acyl CoA in presence of enzyme N-acyltransferase.
The reaction is summarized below.

where R’ = -CH2– (if glycine) or >CH-CH2-CH2-CONH2 (if glutamine)
- Examples of drugs forming glycine or glutamine conjugates are:
- Aliphatic acids e.g. isopropoxyacetic acid
- Alicyclic acids e.g. cholic acid
- Aryl acids e.g. salicylic acid
- Arylacetic acids e.g. phenylacetic acid
- Heterocyclic aryl acids e.g. nicotinic acid.
Conjugation With Glutathione
- Glutathione is a tripeptide with a strongly nucleophilic character due to the presence of a -SH (thiol) group in its structure.
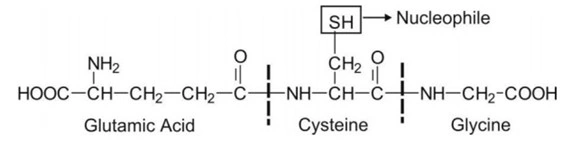
- Thus, it has great affinity for electrophilic substrates, a number of which are potentially toxic compounds. It is important to note that a highly electrophilic metabolite has a tendency to react with tissue nucleophilic groups such as -OH, -NH2 and -SH and precipitate toxicities such as tissue necrosis, carcinogenesis, mutagenesis, teratogenesis, etc. Conjugation with glutathione protects the tissue from such reactive moieties and thus, the reaction is an important detoxication route.
- GSH conjugation differs from other conjugation reactions in that the process does not require initial activation of the coenzyme or the substrate since the GSH, which is anucleophile itself, is highly reactive towards an electrophilic substrate.
- The interaction between the substrate and the GSH is catalysed by enzyme glutathione-S-transferase to form S-substituted glutathione conjugate.
Acetylation
- The general sequence of acetylation reaction is same as that of amino acid conjugation. In acetylation reaction the acetyl group is attached to drug/ metabolite. The enzyme involved is the N-acetyl transferase.
- The drugs/metabolites containing primary amino groups can undergo acetylation. Alcohols (e.g. choline) and thiols (e.g. CoASH) also undergo acetylation with minor extent.
- Acetylation may lead to toxic metabolite, e.g. acetyl derivatives of sulfonamides cause renal toxicity due to reduced water solubility of the metabolites.
Methylation
- It is intermediate reaction between phase I and phase II reactions. The mechanism of methylation reaction is as follow:
- Synthesis of donor of methyl group i.e. activated coenzyme S-Adenosyl Methionine (SAM) from L.-methionine and ATP.
- Transfer of the methyl group from SAM to the drug/metabolite. This reaction is catalyzed by nonmicrosomal enzyme methyl transferase.
- Various methyl transferases responsible for methylation of drug/metabolite are Catechol-O-Methyl Transferase (COMT); Phenyl-O-Methyl Transferase (POMT), Phenyl Ethanolamine-N-Methyl Transferase (PNMT) and nonspecific transferases.
Miscellaneous Conjugation Reactions
Some of the rare conjugation reactions are mentioned below.
Conjugation of Cyanide
Conjugation with Ribose
Conjugation with Taurine
Excretion of Drugs
- Drugs and/or their metabolites are removed from the body by excretion.
- Excretion is defined as the process whereby drugs and/or their metabolites are irreversibly transferred from internal to external environment.
- Excretion of unchanged or intact drug is important in thetermination of its pharmacological action.
- The principal organs of excretion are kidneys. Excretion of drug by kidneys is called as renal excretion. Excretion by organs other than kidneys such as lungs, biliary system, intestine, salivary glands and sweat glands is known as nonrenal excretion.
RENAL EXCRETION OF DRUGS
- Almost all drugs and their metabolites are excreted by the kidneys to some extent or the other. Some drugs such as gentamicin are exclusively eliminated by renal route only.
- Agents that are excreted in urine are –
- Water-soluble.
- Non-volatile.
- Small in molecular size (less than 500 Daltons).
- The ones that are metabolised slowly.
- Nephron: The basic functional unit of kidney involved in excretion is the nephron. Each kidney comprises of one million nephrons. Each nephron is made up of the glomerulus, the proximal tubule, the loop of Henle, the distal tubule and the collecting tubule.
- The principal processes that determine the urinary excretion of a drug are –
- Glomerular filtration.
- Active tubular secretion.
- Active or passive tubular reabsorption.
- These processes are depicted in Fig.3.
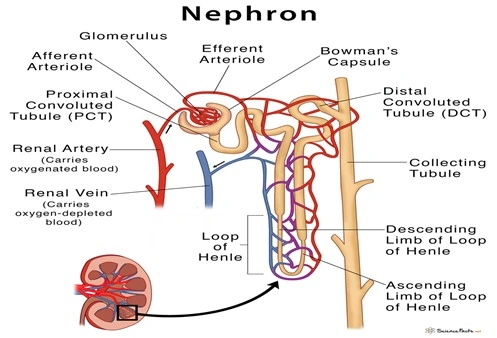
Fig. 2Nephron (information purpose)
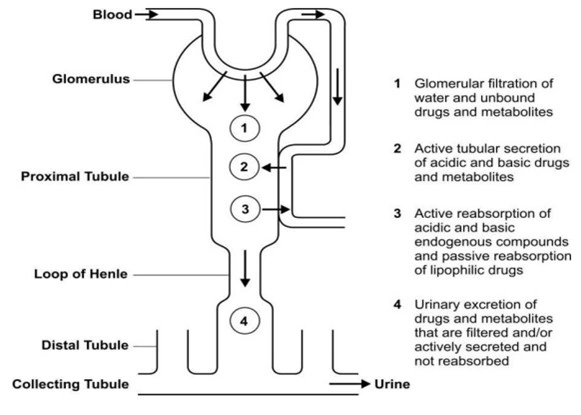
Fig. 3A simplified diagram illustrating processes involved in the urinary excretion ofdrugs
- Glomerular filtration and active tubular secretion tend to increase the concentration of drugs in lumen and hence facilitate excretion whereas tubular reabsorption decreases it and prevents the movement of drug out of the body. Thus, the rate of excretion can be given by equation:
Rate of Excretion = Rate of Filtration + Rate of Secretion – Rate of Reabsorption (Eqn. 1)
Glomerular Filtration
- Glomerular filtration is a non-selective, unidirectional process whereby most compounds, ionised or unionised, are filtered except those that are bound to plasma proteins or blood cells and thus behave as macromolecules.
- The glomerulus also acts as a negatively charged selective barrier promoting retention of anionic compounds. The driving force for filtration through the glomerulus is the hydrostatic pressure of the blood flowing in the capillaries.
- Out of the 25% of cardiac output or 1.2 litres of blood/min that goes to the kidneys via renal artery, only 10% or 120 to 130 ml/min is filtered through the glomeruli, the rate being called as the glomerular filtration rate (GFR).
- Though some 180 litres of protein and cell free ultrafiltrate pass through the glomeruli each day, only about 1.5 litres is excreted as urine, the remainder being reabsorbed from the tubules.
- The GFR can be determined by an agent that is excreted exclusively by filtration and is neither secreted nor reabsorbed in the tubules. The excretion rate value of such an agent is 120 to 130 ml/min. Creatinine, inulin, mannitol and sodium thiosulphate are used to estimate GFR of which the former two are widely used to estimate renal function.
Active Tubular Secretion
- It is a carrier-mediated process which requires energy for transportation of compounds against the concentration gradient.
- The system is capacity-limited and saturable. Two active tubular secretion mechanisms have been identified:
- System for secretion of organic acids/anions like penicillins, salicylates,glucuronides, sulphates, etc. It is the same system by which endogenous acids such as uric acid are secreted.
- System for secretion of organic bases/cations like morphine, mecamylamine,hexamethonium and endogenous amines such as catecholamines, choline, histamine, etc.
- Both the systems are relatively non-selective and independent of each other but both can be bidirectional i.e. agents may both be secreted as well as reabsorbed actively, for example, uric acid.
- Two structurally similar drugs having similar ionic charge and employing the same carrier-mediated process for excretion enter into competition. A drug with greater rate of clearance will retard the excretion of the other drug with which it competes. The half-life of both the drugs is increased since the total sites for active secretion are limited. This may result in accumulation of drugs and thus, precipitation of toxicity. However, the principle of competition can be exploited for therapeutic benefits.
- An interesting example of this is the anionic agent probenecid. Probenecid inhibits the active tubular secretion of penicillins thus increasing their concentration in plasma by at least two fold. A 50% reduction in penicillin G dose is suggested, when it is given with probencid.
Tubular Reabsorption
- Tubular reabsorption occurs after the glomerular filtration of drugs. It takes place all along the renal tubule. Reabsorption of a drug is indicated when the excretion rate values are less than the GFR of 130 ml/min. An agent such as glucose that is completely reabsorbed after filtration has a clearance value of zero. Contrary to tubular secretion, reabsorption results in an increase in the half-life of a drug.
- Tubular reabsorption can either be an:
- Active process, or
- Passive process.
- Active tubular reabsorption is commonly seen with high threshold endogenoussubstances or nutrients that the body needs to conserve such as electrolytes, glucose, vitamins, amino acids, etc. Uric acid is also actively reabsorbed (inhibited by the uricosuric agents). Very few drugs are known to undergo reabsorption actively e.g. oxopurinol.
- Passive tubular reabsorption is common for a large number of exogenous substancesincluding drugs. The driving force for such a process i.e. the concentration gradient is established by the back diffusion or reabsorption of water along with sodium and other inorganic ions.
- Understandably, if a drug is neither secreted nor reabsorbed, its concentration in the urine will be 100 times that of free drug in plasma due to water reabsorption since less than 1% of glomerular filtrate is excreted as urine.
- The primary determinant in the passive reabsorption of drugs is their lipophilicity. Lipophilic substances are extensively reabsorbed while polar molecules are not.
CONCEPT OF CLEARANCE
- The clearance concept was first introduced to describe renal excretion of endogenous compounds in order to measure the kidney function.
- The term is now applied to all organs involved in drug elimination such as liver, lungs, the biliary system, etc. and referred to as hepatic clearance, pulmonary clearance, biliary clearance and so on.
- The sum of individual clearances by all eliminating organs is called as total body clearanceortotal systemicclearance. It is sometimes expressed as a sum of renal clearance and nonrenal clearance.
- Clearance is defined as the theoretical volume of body fluids containing drug from which the drug is removed or cleared completely in a specific period of time. It is expressedin ml/min.
- In comparison to apparent volume of distribution which relates amount of drug in the body to the plasma drug concentration, clearance relates rate of drug elimination to the plasma concentration.

- Renal Clearance (ClR): It can be defined as the volume of blood or plasma which is completely cleared of the unchanged drug by the kidney per unit time. It is expressed mathematically as:

- Physiologically speaking, renal clearance is the ratio of “sum of rate of glomerular filtration and active secretion minus rate of reabsorption” to “plasma drug concentration C”.

FACTORS AFFECTING RENAL EXCRETION OR RENAL CLEARANCE
Factors influencing renal clearance of drugs and metabolites are:
- Physicochemical properties of the drug
- Plasma concentration of the drug
- Distribution and binding characteristics of the drug
- Blood flow to the kidneys
- Biological factors
- Drug interactions
- Disease states
1. Physicochemical Properties of the Drug
- Important physicochemical factors affecting renal excretion of a drug are – molecular size, pKa and lipid solubility.
- The molecular weight of a drug is very critical in its urinary elimination. Drugs of small molecular size with less than 300 Daltons can be easily filtered through the glomerulus and are readily excreted by the kidneys. Drugs in the molecular weight range 300 to 500 Daltons can be excreted both in urine and bile. Molecules of size greater than 500 Daltons are excreted in urine to a lesser extent and excreted through bile.
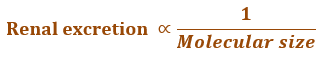
- The influence of drug pKa and lipophilicity on excretion: Urinary excretion of an unchanged drug is inversely related to its lipophilicity. This is because, a lipophilic drug is passively reabsorbed to a large extent.
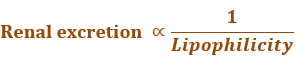
2. Plasma Concentration of the Drug
- Glomerular filtration and reabsorption are directly affected by plasma drug concentration since both are passive processes.
Renal excretion ∝ Plasma concentration of drug
- A drug that is not bound to plasma proteins and excreted by filtration only, shows a linear relationship between rate of excretion and plasma drug concentration.
- In case of drugs which are secreted or reabsorbed actively, the rate process increases with an increase in plasma concentration to a point when saturation of carrier occurs. In case of actively reabsorbed drugs, excretion is negligible at low plasma concentrations. Such agents are excreted in urine only when their concentration in the glomerular filtrate exceeds the active reabsorption capacity, e.g. glucose. With drugs that are actively secreted, the rate of excretion increases with increase in plasma concentration up to a saturation level. These situations are depicted in Fig. 4.
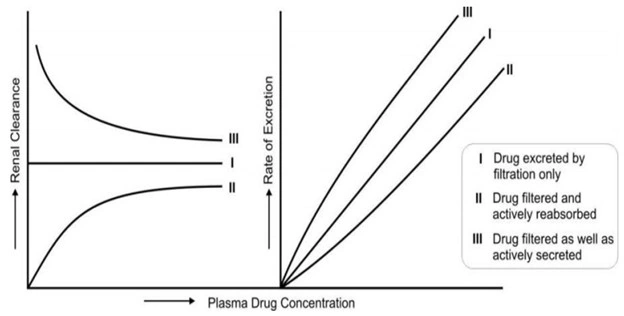
Fig. 4. Renal clearance and rate of excretion of a drug in relation to its plasma concentration as affected by the physiological processes – filtration, active reabsorption and active secretion.
3. Distribution and Binding Characteristics of the Drug
- Clearance is inversely related to apparent volume of distribution of drugs. A drug with large Vd is poorly excreted in urine. Drugs restricted to blood compartment have higher excretion rates.

- Drugs that are bound to plasma proteins behave as macromolecules and thus cannot be filtered through the glomerulus. Only unbound or free drug appear in the glomerular filtrate.
- Drugs extensively bound to proteins have long half-lives because the renal clearance is small and urine flow rate is just 1 to 2 ml/min. The renal clearance of oxytetracycline which is 66% unbound is 99 ml/min while that of doxycycline (7% unbound) is just 16 ml/min.
- Actively secreted drugs are much less affected by protein binding, e.g. penicillins. The free fraction of such drugs are filtered as well as secreted actively and dissociation of drug-protein complex occurs rapidly.
4. Blood Flow to the Kidneys
- The renal blood flow is important in case of drugs excreted by glomerular filtration only and those that are actively secreted. In the latter case, increased perfusion increases the contact of drug with the secretory sites and enhances their elimination.
Renal excretion ∝ Blood flow to kidney
5. Biological Factors
- Age, sex, species and strain differences, differences in the genetic make-up, circadian rhythm, etc. alter drug excretion.
- Renal excretion is approximately 10% lower in females than in males.
- The renal function of newborns is 30 to 40% less in comparison to adults and attains maturity between 2.5 to 5 months of age.
- In old age, the GFR is reduced and tubular function is altered, the excretion of drugs is thus slowed down and half-life is prolonged.
6. Drug Interactions
- Any drug interaction that results in alteration of protein-drug binding characteristics, renal flood flow, active secretion, urine pH and intrinsic clearance and forced diuresis would alter renal clearance of a drug.
- Alteration of Urine pH: Acidification of urine with ammonium chloride, methionineor ascorbic acid enhances excretion of basic drugs.
Alkalinisation of urine with citrates, tartarates, bicarbonates and carbonic anhydrase inhibitors enhances excretion of acidic drugs. - Competition for Active Secretion: the drug which utilize the same carrier for active tubular secretion, can compete with each other for binding with carrier which affects excretion.
- Forced Diuresis: All diuretics increase elimination of drugs whose renal clearancegets affected by urine flow rate.
7. Disease States—Renal Impairment
- Renal dysfunction greatly impairs the elimination of drugs especially those that areprimarily excreted by the kidneys. Some of the causes of renal failure are hypertension, diabetes mellitus, hypovolemia (decreased blood supply to the kidneys), pyelonephritis (inflammation of kidney due to infections, etc.), nephroallergens (e.g. nephrotoxic serum) and nephrotoxic agents such as aminoglycosides, phenacetin and heavy metals such as lead and mercury.
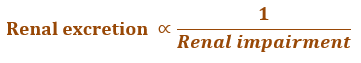
- Uraemia, characterized by impaired glomerular filtration and accumulation of fluids andprotein metabolites, also impairs renal clearance of drugs. In both these conditions, the half-lives of drugs are increased. As a consequence, drug accumulation and toxicity may result. Determination of renal function is therefore important in such conditions in order to monitor the dosage regimen.
NON-RENAL ROUTES OF DRUG EXCRETION
Drugs and their metabolites may also be excreted by routes other than the renal route, called as the extrarenal ornonrenal routes of drug excretion. The various such excretionprocesses are:
- Biliary excretion (Liver-Faeces)
- Pulmonary excretion (Lungs)
- Salivary excretion
- Mammary excretion (Breast milk)
- Skin/dermal excretion
- Gastrointestinal excretion
- Genital excretion (Reproductive organ)
1. Biliary Excretion of Drugs
- Bile is produced by the hepatic cells lining the bile canaliculi.
- It is produced in liver and transported from the liver through the hepatic ducts, stored and concentrated in the gallbladder, and released into the duodenum (the first part of the small intestine) via the common bile duct, stimulated by meals.
- In humans, the bile flow rate is steady 0.5 to 1 mL/min. Almost 90% of the secreted bile acids are reabsorbed from the intestine and transported back to the liver for re-secretion. The rest is excreted in faeces.
- A drug, whose biliary concentration is less than that in plasma, has a small biliary clearance and vice versa.
- A higher bile/plasma concentration ratio generally indicates more excretion of a compound into the bile.
- Compounds that are excreted in bile have been classified into 3 categories on the basis of their bile/plasma concentration ratios:
- Group A compounds whose ratio is approximately 1, e.g. sodium, potassium and chloride ions and glucose.
- Group B compounds whose ratio is >1, usually from 10 to 1000, e.g. bile salts, bilirubinglucuronide, creatinine, sulphobromophthalein conjugates, etc.
- Group C compounds with ratio < 1, e.g. sucrose, inulin, phosphates, phospholipids andmucoproteins.
- Several factors influence secretion of drugs in bile –
- The most important factor governing the excretion of drugs in bile is their molecular weight. Its influence on biliary excretion is summarized in the below Table.
- Polarity is the other physicochemical property of drug influencing biliary excretion. Greater the polarity, better the excretion. Thus, metabolites are more excreted in bile than the parent drugs because of their increased polarity.

- Orally administered drugs which during absorption process go to the liver, are excreted more in bile in comparison to parenterally administered drugs.
- The ability of liver to excrete the drug in the bile is expressed by biliary clearance.

2. Pulmonary Excretion
- Pulmonary excretion is the process by which drugs, specifically gaseous and volatile substances, are eliminated from the body through exhaled air.
- Gaseous and volatile substances such as the general anaesthetics (e.g. halothane) are absorbed through the lungs by simple diffusion. Similarly, their excretion by diffusion into the expired air is possible.
- Factors influencing pulmonary excretion of a drug include pulmonary blood flow, rate of respiration, solubility of the volatile substance, etc.
- Cardiac Output: High blood flow to the lungs increases the amount of drug delivered for excretion.
- Respiration Rate: Faster breathing speeds up the removal of the drug from the alveolar space, maintaining the concentration gradient.
- Compounds like alcohol which have high solubility in blood and tissues are excreted slowly by the lungs.
3. Salivary Excretion
- Salivary excretion is a minor route of drug elimination where substances pass from the blood plasma into the saliva.
- Most drugs enter saliva through passive diffusion across the epithelial cells of the salivary glands.
- Selective Entry: Only the unbound, lipid-soluble, and unionized form of a drug can easily cross these membranes.
- Active Transport: Some drugs, such as metformin and penicillin are transported into saliva via specialized proteins like the organic cation transporter OCT3.
- Recycling: Because saliva is typically swallowed, many excreted drugs are reabsorbed in the gastrointestinal tract, leading to a form of internal recycling rather than permanent elimination from the body.
- Basic drugs are excreted more in saliva as compared to acidic drugs.
- Drug excreted in saliva, e.g. Penicillin, caffeine, theophylline, phenytoin, carbamazepine, etc.
- The bitter after taste in the mouth of a patient on medication is an indication of drug excretion in Saliva.
4. Mammary Excretion (Breast milk)
- Mammary excretion is the process by which drugs and their metabolites pass from a mother’s systemic circulation into breast milk. While it is a minor route for the mother’s elimination.
- Excretion of a drug in milk is clinically critical/ important since it can gain entry into the breast-feeding infant.
- Drugs enter breast milk primarily through passive diffusion across the epithelial membrane of the mammary glands.
- The “Blood-Milk Barrier”: During the first few days after birth (the colostrum phase), the gaps between mammary cells are large, allowing almost all drugs to pass easily. As the milk matures, these gaps close, making the barrier more selective.
- Lipid Solubility: Since breast milk has a higher fat content than plasma, lipid-soluble (lipophilic) drugs (like diazepam) concentrate more easily in milk.
- Since milk is acidic (6.6 to 7.4) in comparison to plasma (7.0 to 7.2), weakly basic drugs concentrate more in milk.
- Drugs extensively bound to plasma proteins, e.g. diazepam, are less secreted in milk.
- The amount of drug excreted in milk is generally less than 1% and the fraction consumed by the infant is too less to reach therapeutic or toxic levels. But some potent drugs such as barbiturates, morphine and ergotamine may induce toxicity in infants. Some examples of toxicity to breast-fed infants owing to excretion of drug in milk are –
- Chloramphenicol: Possible bone marrow suppression.
- Diazepam: Accumulation and sedation.
- Heroin: Prolonged neonatal dependence.
- Methadone: Possible withdrawal syndrome if breast-feeding is stopped suddenly.
- Propylthiouracil: Suppression of thyroid function.
- Tetracycline: Permanent staining of infant teeth.
- Wherever possible, nursing mothers should avoid drugs. If medication is unavoidable, the infant should be bottle-fed.
5. Skin Excretion
- Skin excretion is a minor route of drug elimination where substances are removed from the body through sweat or sebum.
- Drugs reach the skin surface through two main glands:
- Eccrine Sweat Glands: Most drugs enter sweat via passive diffusion from the blood capillaries surrounding the glands. Only the unbound (free) and unionized form of a drug typically crosses into sweat.
- Sebaceous Glands: These glands secrete sebum (skin oil). Highly lipid-soluble (lipophilic) drugs tend to accumulate here, as sebum is rich in fats.
- Factors Influencing Skin Excretion
- pH Partitioning: Sweat is typically more acidic (pH ~4.0 to 6.8) than blood (pH 7.4). This can cause basic drugs (like amphetamines) to become “trapped” in sweat at higher concentrations than in the blood.
- Lipid Solubility: Since the skin surface and sebum are fatty, drugs with high lipid solubility (like griseofulvin or ketoconazole) are excreted more efficiently this way.
- Sweat Rate: Physical activity, high ambient temperature, or certain fevers increase sweat production, which can accelerate the excretion of water-soluble drugs like urea or heavy metals.
6. Gastrointestinal Excretion
- Excretion of drugs into the GIT usually occurs after parenteral administration when the concentration gradient for passive diffusion is favourable. The process is reverse of GI absorption of drugs.
- Water soluble and ionised form of weakly acidic and basic drugs is excreted in the GIT, e.g. nicotine and quinine are excreted in to stomach. Orally administered drugs can also be absorbed and excreted in the GIT.
- Drugs excreted in the GIT are reabsorbed into the systemic circulation and undergo recycling.
7. Genital Excretion
- Genital excretion refers to the elimination of drugs or their metabolites through the secretions of the male or female reproductive tracts, specifically semen (seminal fluid) and vaginal secretions.
- Drugs enter genital secretions primarily via passive diffusion from the blood plasma across the epithelial barriers of the prostate, seminal vesicles, or vaginal wall.
- The Blood-Testis/Blood-Prostate Barrier: These are tight junctions that limit the passage of many drugs. Only lipid-soluble, unbound, and unionized drugs can easily cross.
- Vaginal fluid is also acidic (pH ~4.0–4.5), which can similarly influence the concentration of basic drugs.
A summary of drugs excreted by various routes is given in below Table.
| Excretory Route | Mechanism | Characteristics of Drugs Excreted |
| Renal excretion | Glomerular filtration, active secretion, active/passive reabsorption | Free, hydrophilic, unchanged drugs/metabolites/conjugates with Molecular Weight (MW) < 500. |
| Biliary excretion (Liver-Faeces) | Active secretion | Hydrophilic, unchanged drugs/metabolites/conjugates with MW ≥ 500. |
| Pulmonary excretion (Lungs) | Passive diffusion | Gaseous and volatile substances; drugs that are insoluble in blood and tissue. |
| Salivary excretion | Passive diffusion, active transport | Free, unionised, lipophilic drugs; some polar drugs. |
| Mammary excretion (Breast milk) | Passive diffusion | Free, unionised, lipophilic drugs (generally basic in nature). |
| Skin/dermal excretion (Sweat) | Passive diffusion | Free, unionised, lipophilic drugs. |
| Gastrointestinal excretion | Passive diffusion | Water-soluble, ionised drugs. |
| Genital excretion (Reproductive organ) | Passive diffusion | lipid-soluble, unbound, basic (vegina) and unionized drugs |
Bioavailability and Bioequivalence
- Bioavailability is defined as the rate and extent (amount) of absorption of unchanged drug from its dosage form into the systemic circulation.
- Bioequivalence is a comparative term. Two drug products are considered bioequivalent if their bioavailabilities do not show a significant difference when administered at the same dose under similar conditions.
Bioavailability:
- The therapeutic effectiveness of a drug depends upon the ability of the dosage form to deliver the medicament to its site of action at a rate and amount sufficient to elicit the desired pharmacological response. This attribute of the dosage form is referred to as physiological availability, biological availability or simply, bioavailability.
- For mostdrugs, the pharmacological response can be related directly to the plasma levels. Thus, the term bioavailability is defined as the rate and extent (amount) of absorption of unchanged drug from its dosage form into the systemic circulation.
- The rate or rapidity withwhich a drug is absorbed is an important consideration when a rapid onset of action is desired as in the treatment of acute conditions such as asthma attack, pain, etc. A slower absorption rate is, however, desired when the aim is to prolong the duration of action or to avoid the adverse effects. On the other hand, extent of absorption is of special significance in the treatment of chronic conditions like hypertension, epilepsy, etc.
- If the size of the dose to be administered is same, then bioavailability of a drug from its dosage form depends upon 3 major factors:
- Pharmaceutical factors related to physicochemical properties of the drug andcharacteristics of the dosage form.
- Patient-related factors.
- Route of administration.
- The former two factors (pharmaceutical factors and patient-related factors) have been dealt with comprehensively in chapter–Absorption of Drugs.
- The influence of route of administration on drug’s bioavailability isgenerally in the following order with few exceptions:
Parenteral > Oral > Rectal > Topical
- Within the parenteral route, intravenous injection of a drug results in 100% bioavailability as the absorption process is bypassed. However, for reasons of stability and convenience, most drugs are administered orally. In such cases, the dose available to the patient, called as the bioavailable dose, is often less than the administered dose.
- The amount of drug that reaches the systemic circulation (i.e. extent of absorption) is called as systemic availabilityor simplyavailability.
- The term bioavailable fraction F, refers to the fraction of administered dose that enters the systemic circulation.

Objectives of Bioavailability Studies
Bioavailability studies are important in the —
- Primary stages of development of a suitable dosage form for a new drug entity to obtain evidence of its therapeutic utility.
- Development of new formulations of the existing drugs.
- Determination of influence of excipients, patient related factors and possible interaction with other drugs on the efficiency of absorption.
- Control of quality of a drug product during the early stages of marketing in order to determine the influence of processing factors, storage and stability on drug absorption.
- Comparison of availability of a drug substance from different dosage forms or from the same dosage form produced by different manufacturers.
Bioavailability-Absolute versus Relative
Absolute Bioavailability
- When the systemic availability of a drug administered orally is determined in comparison to its intravenous administration, it is called as absolute bioavailability. It is denoted by symbol F.
- Its determination is used to characterize a drug ‘s inherent absorption properties from the extravascular site.
- Intravenous dose is selected as a standard because the drug is administered directly into the systemic circulation and it shows 100% bioavailability and avoids absorption step.
- Intramuscular dose can also be taken as a standard if the drug is poorly water-soluble.
- It is denoted by symbol F
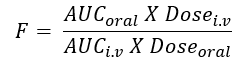
Relative Bioavailability
- When the systemic availability of a drug after oral administration is compared with that of an oral standard of the same drug – such as an aqueous or non-aqueous solution ora suspension, it is referred to as relative or comparative bioavailability.
- It is denoted by symbol Fr. In contrast to absolute bioavailability, it is used to characterize absorption of a drug from its formulation. F and Fr are generally expressed in percentages.
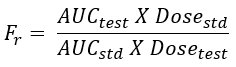
Measurement of Bioavailability
- The systemic bioavailability of drug from dosage form is measured using two techniques i.e., single dose study and multiple dose study.
- Single dose studies are very common and carried out by administrating single dose of drug in test subjects. These are convenient, less expensive and require less exposure of drugs to test subjects. However, the steady-state absorption characteristics and inter-subject variability of drug are difficult to predict with such studies.
- The multiple dose studies are difficult to control, and more amount of drug is expose to test subjects. However steady-state absorption characteristics and inter-subject variability of drug can be monitored using these studies. The dose of drug to be administered for a bioavailability study is selected based on preliminary clinical experiments.
The methods useful in quantitative evaluation of bioavailability can be broadly divided into two categories — I. pharmacokinetic methods and II. pharmacodynamic methods.
I. Pharmacokinetic Methods
These are very widely used and based on the assumption that the pharmacokinetic profile reflects the therapeutic effectiveness of a drug. Thus, these are indirect methods. The two major pharmacokinetic methods are:
1. Plasma level-time studies.
2. Urinary excretion studies.
II. Pharmacodynamic Methods
These methods are complementary to pharmacokinetic approaches and involve direct measurement of drug effect on a (patho)physiological process as a function of time. The two pharmacodynamic methods involve determination of bioavailability from:
1. Acute pharmacological response.
2. Therapeutic response.
1. Plasma Level—Time Studies
- Unless determination of plasma drug concentration is difficult or impossible, it is the most reliable method and method of choice in comparison to urine data. The method is based on the assumption that two dosage forms that exhibit superimposable and identical plasma level-time profiles in a group of subjects should result in identical therapeutic activity.
- With single dose study, the method requires collection of serial blood samples for a period of 2 to 3 biological half-lives after drug administration, their analysis for drug concentration and making a plot of concentration versus corresponding time of sample collection to obtain the plasma level-time profile.
- With i.v. dose, sampling should start within 5 minutes of drug administration and subsequent samples taken at 15-minute intervals. To adequately describe the disposition phase, at least 3 sample points should be taken if the drug follows one-compartment kinetics and 5 to 6 points if it fits two-compartment model.
- For oral dose, at least 3 points should be taken on the ascending part of the curve for accurate determination of Ka (absorption rate constant). The points for disposition or descending phase of the curve must be taken in a manner similar to that for i.v. dose.
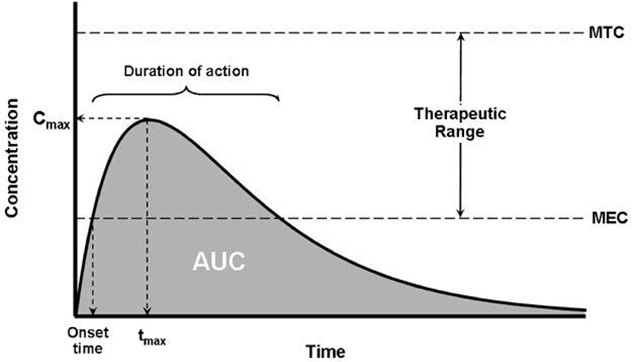
- The 3 parameters of plasma level-time studies which are considered important for determining bioavailability are:
- Cmax: The peak plasma concentration that gives an indication whether thedrug is sufficiently absorbed systemically to provide a therapeutic response. It is a function of both the rate and extent of absorption. Cmax will increase with an increase in the dose, as well as with an increase in the absorption rate.
- tmax: The peak time that gives an indication of the rate of absorption. Itdecreases as the rate of absorption increases.
- AUC: The area under the plasma level-time curve that gives a measure of the extent of absorption or the amount of drug that reaches the systemic circulation.
- The extent of bioavailability can be determined by following equations:
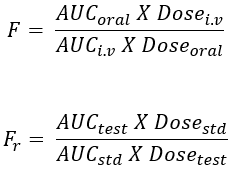
Where, Dose stands for dose administered and subscripts iv and oral indicate the route of administration. Subscripts test and std indicate the test and the standard doses of the same drug to determine relative availability.
- With multiple dose study, the method involves drug administration for at least 5 biological half-lives with a dosing interval equal to or greater than the biological half-life (i.e. administration of at least 5 doses) to reach the steady-state.
- A blood sample should be taken at the end of previous dosing interval and 8 to 10 samples after the administration of next dose. The extent of bioavailability is given as:
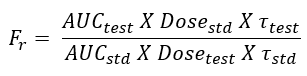
where [AUC] values are area under the plasma level-time curve of one dosing interval in a multiple dosage regimen, after reaching the steady-state (Fig. 6) and τ is the dosing interval.
- Bioavailability can also be determined from the peak plasma concentration at steady-state Css,max according to following equation:
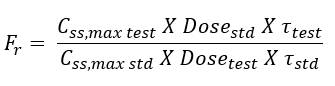
- The rate of absorption is not important in the multiple dosing methods.
2. Urinary Excretion Studies
- This method of assessing bioavailability is based on the principle that the urinary excretion of unchanged drug is directly proportional to the plasma concentration of drug.As a rule of thumb, determination of bioavailability using urinary excretion data should be conducted only if at least 20% of administered dose is excreted unchanged in the urine.
- The study is particularly useful for –
- Drugs extensively excreted unchanged in the urine – for example, certain thiazide diuretics and sulphonamides.
- Drugs that have urine as the site of action – for example, urinary antiseptics such as nitrofurantoin and hexamine.
- Concentration of metabolites excreted in urine is never taken into account in calculations. The method involves –
- Collection of urine at regular intervals for a time-span equal to 7 biological half-lives.
- Analysis of unchanged drug in the collected sample.
- Determination of the amount of drug excreted in each interval and cumulative amount excreted.
- At each sample collection, total emptying of the bladder is necessary to avoid errors resulting from addition of residual amount to the next urine sample.
- The three major parameters examined in urinary excretion data obtained with a single dose study are:
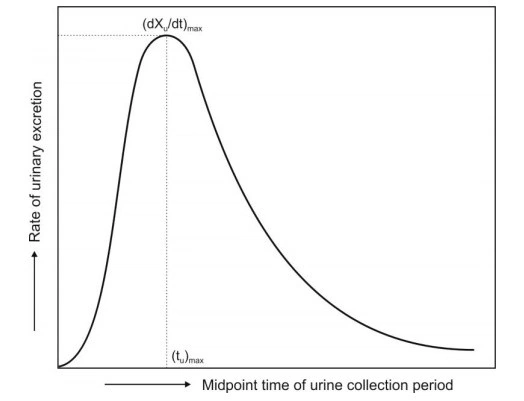
- (dXu/dt)max: The maximum urinary excretion rate, it is obtained from thepeak of plot between rate of excretion versus midpoint time of urine collection period. It is analogous to the Cmax derived from plasma level studies since the rate of appearance of drug in the urine is proportional to its concentration in systemic circulation. Its value increases as the rate of and/or extent of absorption increases (see Fig. 7).
- (tu)max: The time for maximum excretion rate, it is analogous to thetmaxofplasma level data. Its value decreases as the absorption rate increases.
- Xu ∞ : The cumulative amount of drug excreted in the urine, it is related to the AUC of plasma level data and increases as the extent of absorption increases.
- With single dose, the extent of bioavailability is calculated from equations given below:
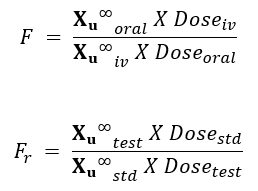
- With multiple dose study to steady-state, the equation for computing bioavailability is:
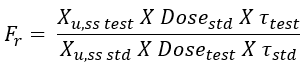
where (Xu,ss) is the amount of drug excreted unchanged during a single dosing interval at steady-state.
- In practice, estimation of bioavailability by urinary excretion method is subject to a high degree of variability, and is less reliable than those obtained from plasma concentration-time profiles. It is thus not recommended as a substitute for blood concentration data; rather, it should be used in conjunction with blood level data for confirmatory purposes.
3. Acute Pharmacological Response Method
- When bioavailability measurement by pharmacokinetic methods is difficult, inaccurate or non-reproducible, an acute pharmacological effect such as a change in ECG or EEG readings, pupil diameter, etc. is related to the time course of a given drug.
- Bioavailability can then be determined by construction of pharmacological effect-time curve as well as dose-response graphs. The method requires measurement of responses for at least 3 biological half-lives of the drug in order to obtain a good estimate of AUC.
- Disadvantages of this method include–
- The pharmacological response tends to be more variable and accurate correlation between measured response and drug available from the formulation is difficult.
- The observed response may be due to an active metabolite whose concentration is not proportional to the concentration of parent drug responsible for the pharmacological effect.
4. Therapeutic Response Method
- Theoretically the most definite, this method is based on observing the clinical response to a drug formulation given to patients suffering from disease for which it is intended to be used. However, the method has several drawbacks –
- Quantitation of observed response is too improper to allow for reasonable assessment of relative bioavailability between two dosage forms of the same drug.
- Unless multiple-dose protocols are employed, a patient who required the drug for a disease would be able to receive only a single dose of the drug every few days or perhaps each week.
- Many patients receive more than one drug, and the results obtained from a bioavailability study could be compromised because of a drug–drug interaction.
In Vitro Drug Dissolution Testing Models

- There are several factors that must be considered in the design of a dissolution test. They are –
- Factors relating to the dissolution apparatus such as—the design, the size of thecontainer (several mL to several litres), the shape of the container (round bottomed or flat), nature of agitation (stirring, rotating or oscillating methods), speed of agitation, performance precision of the apparatus, etc.
- Factors relating to the dissolution fluid such as—composition (water, 0.1N HCl,phosphate buffer, simulated gastric fluid, simulated intestinal fluid, etc.), viscosity, volume (generally larger than that needed to completely dissolve the drug under test), temperature (generally 37oC) and maintenance of sink (drug concentration in solution maintained constant at a low level) or non-sink conditions (gradual increase in the drug concentration in the dissolution medium).
- Process parameters such as method of introduction of dosage form, samplingtechniques, changing the dissolution fluid, etc.
The ideal features of a dissolution apparatus are:
- The fabrication, dimensions, and positioning of all components must be precisely specified and reproducible: run-to-run.
- The apparatus must be simple in design, easy to operate and useable under a variety of conditions.
- The apparatus must be sensitive enough to reveal process changes and formulation differences but still yield repeatable results under identical conditions.
- The apparatus, in most cases, should permit controlled variable intensity of mild, uniform, non-turbulent liquid agitation.
- Nearly perfect sink conditions should be maintained.
- The apparatus should provide an easy means of introducing the dosage form into the dissolution medium and holding it, once immersed, in a regular reliable fashion.
- The apparatus should provide minimum mechanical abrasion to the dosage form during the test period to avoid disruption of the microenvironment surrounding the dissolving form.
- Evaporation of the solvent medium must be eliminated, and the medium must be maintained at a fixed temperature within a specified narrow range. Most apparatuses are thermostatically controlled at around 37°C.
- Samples should be easily withdrawn for automatic or manual analysis without interrupting the flow characteristics of the liquid.
- The apparatus should be capable of allowing the evaluation of disintegrating, non-disintegrating, dense or floating tablets or capsules, and finely powdered drugs.
General Types
The dissolution apparatus has evolved gradually and considerably from a simple beaker type to a highly versatile and fully automated instrument. The devices can be classified in a number of ways. Based on the absence or presence of sink conditions, there are two principal types of dissolution apparatus:
- Closed-compartment apparatus: It is basically a limited-volume apparatusoperating under non-sink conditions. The dissolution fluid is restrained to the size of the container, e.g. beaker type apparatuses such as the rotating basket and the rotating paddle apparatus.
- Open-compartment (continuous flow-through) apparatus: It is the one inwhich the dosage form is contained in a column which is brought in continuous contact with fresh, flowing dissolution medium (perfect sink condition).
USP Types
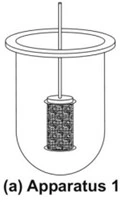
- Rotating Basket Apparatus (Apparatus 1)
- It is basically a closed-compartment, beaker type apparatus comprising of a cylindrical glass vessel with hemispherical bottom of one litre capacity partially immersed in a water bath to maintain the temperature at 37oC.
- A cylindrical basket made of 22 mesh to hold the dosage form is located centrally in the vessel at a distance of 2 cm from the bottom and rotated by a variable speed motor through a shaft (Fig. 8a). The basket should remain in motion during drawing of samples. All metal parts like basket and shaft are made of SS 316.
- Rotating Paddle Apparatus (Apparatus 2)
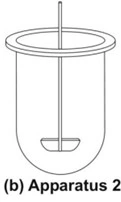
- The assembly is same as that for apparatus 1 except that the rotating basket is replaced with a paddle which acts as a stirrer (Fig. 8b).
- The dosage form is allowed to sink to the bottom of the vessel. Sinkers are recommended to prevent floating of capsules and other floatable forms. A small, loose, wire helix may be attached to such preparations to prevent them from floating.

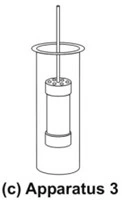
- Reciprocating Cylinder Apparatus (Apparatus 3)
- This apparatus consists of a set of cylindrical flat-bottomed glass vessels equipped with reciprocating cylinders (Fig. 8c). The apparatus is particularly used for dissolution testing of controlled release bead-type (pellet) formulations.
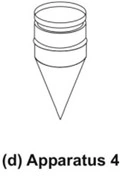
- Flow-Through Cell Apparatus (Apparatus 4)
- The flow-through apparatus consists of a reservoir for the dissolution medium and a pump that forces dissolution medium through the cell holding the test sample. It may be used in either
- Closed-mode where the fluid is recirculated and, by necessity, is of fixed volume, or
- Open-mode when there is continuous replenishment of the fluids.
- The material under test (tablet, capsules, or granules) is placed in the vertically mounted dissolution cell, which permits fresh solvent to be pumped (between 240 and 960 mL/h) in from the bottom (Fig. 8d). Advantages of this apparatus include –
- Easy maintenance of sink conditions for dissolution which is often required for drugs having limited aqueous solubility.
- Feasibility of using large volume of dissolution fluid.
- Feasibility for automation of apparatus.
- The flow-through apparatus consists of a reservoir for the dissolution medium and a pump that forces dissolution medium through the cell holding the test sample. It may be used in either

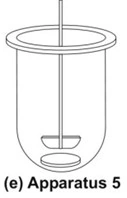
- Paddle Over Disc Apparatus (Apparatus 5)
- This apparatus is used for evaluation of transdermal products and consists of a sample holder or disc that holds the product. The disc is placed at the bottom of apparatus 2 (rotating paddle apparatus; see fig. 8e) and the apparatus operated in the usual way.
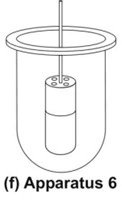
- Cylinder Apparatus (Apparatus 6)
- This apparatus is also used for evaluation of transdermal products and is similar to apparatus 1 (Fig. 8f). Instead of basket, a stainless steel cylinder is used to hold the sample. The sample is mounted on an inert porous cellulosic material and adhered to the cylinder.
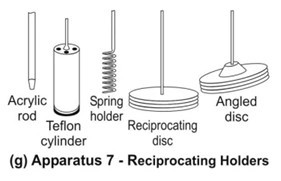
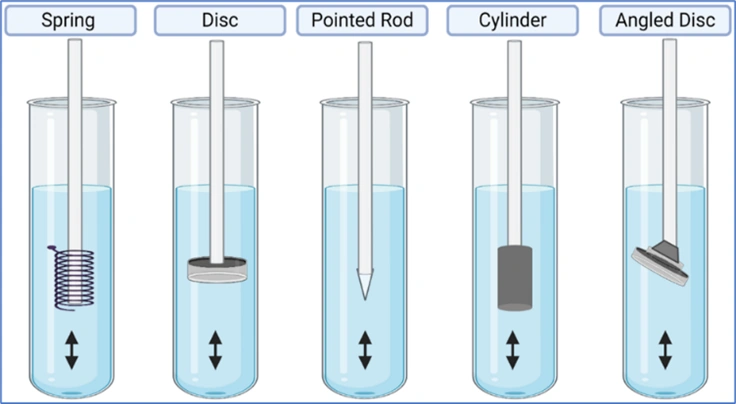
- Reciprocating Disc Apparatus (Apparatus 7)
- This apparatus is used for evaluation of transdermal products as well as non-disintegrating controlled-release oral preparations. The samples are placed on disc-shaped holders (Fig. 8g) using inert porous cellulosic support which reciprocates vertically by means of a drive inside a glass container containing dissolution medium. The test is carried out at 320C and reciprocating frequency of 30 cycles/min.
Compendial Dissolution Apparatus Types and Their Applications
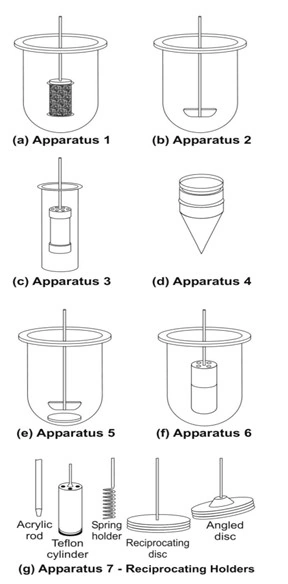
Fig. 8 Schematic representation of official USP dissolution apparatus – (a) Apparatus 1 – rotating basket apparatus, (b) Apparatus 2 – rotating paddle apparatus, (c) Apparatus 3 – reciprocating cylinder apparatus, (d) Apparatus 4 – flow through cell apparatus, (e) Apparatus 5 – paddle over disc apparatus, (f) Apparatus 6 – cylinder apparatus, and (g) Apparatus 7 – reciprocating disc apparatus
| Apparatus | Name | Drug Formulation Tested |
| Apparatus 1 | Rotating basket | Conventional Tablets |
| Apparatus 2 | Rotating paddle | Tablets, capsules, controlled release products, suspensions |
| Apparatus 3 | Reciprocating cylinder | Controlled release formulations |
| Apparatus 4 | Flow-through cell | Formulations containing poorly soluble drugs |
| Apparatus 5 | Paddle over disc | Transdermal formulations |
| Apparatus 6 | Cylinder | Transdermal formulations |
| Apparatus 7 | Reciprocating disc | Controlled release formulations |
In Vitro—In Vivo Correlation (IVIVC)
- A simple in-vitro dissolution test on the drug product will be insufficient to predict its therapeutic efficacy.
- Convincing correlation between in-vitro dissolution behaviour of a drug and its in-vivo bioavailability must be experimentally demonstrated to guarantee reproducibility of biologic response.
- In vitro-in vivo correlation is defined as the predictive mathematical model that describes the relationship between an in-vitro property (such as the rate and extent of dissolution) of a dosage form and an in-vivo response (such as the plasma drug concentration or amount of drug absorbed).
- The main objective of developing and evaluating an IVIVC is to enable the dissolution test to serve as a surrogate (alternate) for in vivo bioavailability studies in human beings.
- The applications of developing such an IVIVC are —
- To ensure batch-to-batch consistency in the physiological performance of a drug product by use of such in vitro values.
- To serve as a tool in the development of a new dosage form with desired in vivo performance.
- To assist in validating or setting dissolution specifications (i.e. the dissolution specifications are based on the performance of product in vivo).
- There are two basic approaches by which a correlation between dissolution testing and bioavailability can be developed:
- By establishing a relationship, usually linear, between the in vitro dissolution and the in vivo bioavailability parameters.
- By using the data from previous bioavailability studies to modify the dissolution methodology in order to arrive at meaningful in vitro-in vivo correlation.
- Some of the often used quantitative linear in vitro-in vivo correlations are –
- Correlations Based on the Plasma Level Data: Here linear relationships between dissolution parameters such as percent drug dissolved, rate of dissolution, rate constant for dissolution, etc. and parameters obtained from plasma level data such as percent drug absorbed, rate of absorption, Cmax, tmax, Ka, etc. are developed; for example, percent drug dissolved versus percent drug absorbed plots.
- Correlation Based on the Urinary Excretion Data: Here, dissolution parameters are correlated to the amount of drug excreted unchanged in the urine, cumulative amount of drug excreted as a function of time, etc.
- Correlation Based on the Pharmacological Response: An acutepharmacological effect such as LD50 in animals is related to any of the dissolution parameters.
In vitro-In vivo Correlation Levels
Three IVIVC levels have been defined and categorised in descending order of usefulness.
- Level A –
- The highest category of correlation, it represents a point-to-point relationship between in vitro dissolution and the in vivo rate of absorption (or in vivo dissolution) i.e. the in vitro dissolution and in vivo absorption rate curves are superimposable and the mathematical description for both curves is the same.
- Advantages of level A correlation is a point-to-point correlation is developed. The in vitro dissolution curve serves as a surrogate for in-vivo performance. Any change in manufacturing procedure or modification in formula can be justified without the need for additional human studies.
- Level B –
- Utilizes the principles of statistical moment analysis. The mean in-vitro dissolution time is compared to the mean in-vivo dissolution time.
- However, such a correlation is not a point-to-point correlation since there are a number of in-vivo curves that will produce similar mean residence time values. It is for this reason that one cannot rely upon level B correlation to justify changes in manufacturing or modification in formula. Moreover, the in vitro data cannot be used for quality control standards.
- Level C –
- It is a single point correlation. It relates one dissolution time point (e.g.t50%, etc.) to one pharmacokinetic parameter such as AUC, tmax or C max. This level is generally useful only as a guide in formulation development or quality control owing to its obvious limitations.
Multiple Level C – It is correlation involving one or several pharmacokinetic parameters to the amount of drug dissolved at various time points.
- It is a single point correlation. It relates one dissolution time point (e.g.t50%, etc.) to one pharmacokinetic parameter such as AUC, tmax or C max. This level is generally useful only as a guide in formulation development or quality control owing to its obvious limitations.
BIOEQUIVALENCE STUDIES
- Bioavailability is defined as the rate and extent (amount) of absorption of unchanged drug from its dosage form into the systemic circulation.
- Bioequivalence is a comparative term. Two drug products are considered bioequivalent if their bioavailabilities does not show a significant difference when administered at the same dose under similar conditions.
Objectives for Biequivalence Studies
- ANDA (Abbreviated New Drug Application): If a new product is intended to be a substitute for an approved medicinal product as a pharmaceutical equivalent or alternative, the equivalence with this product should be shown or justified. In order to ensure clinical performance of such drug products, bioequivalence studies should be performed.
- Evaluation of Post-Approval Changes: Even the original manufacturer must conduct bioequivalence studies if they make significant changes to an already approved drug.
Like: Change in Excipients, Change in Manufacturing Process, Change in Manufacturing Site.
Some of the important terms relevant in this context will be defined.
- Equivalence: It is a relative term that compares drug products with respect to a specific characteristic or function or to a defined set of standards. There are several types of equivalences.
- Chemical Equivalence: It indicates that two or more drug products contain the samelabelled chemical substance as an active ingredient in the same amount.
- Pharmaceutical Equivalence: This term implies that two or more drug products are identical in strength, quality, purity, content uniformity and disintegration and dissolution characteristics; they may however differ in containing different excipients.
- Bioequivalence is a comparative term. Two drug products are considered bioequivalent if their bioavailabilities does not show a significant difference when administered at the same dose under similar conditions i.e., their plasma concentration-time profiles will be identical without significant statistical differences.
When statistically significant differences are observed in the bioavailability of two or more drug products, bio-inequivalence is indicated. - Therapeutic Equivalence: This term indicates that two or more drug products thatcontain the same therapeutically active ingredient elicit identical pharmacological effects and can control the disease to the same extent.
Types of Bioequivalence Studies
Bioequivalence can be demonstrated either –
- In vivo, or
- In vitro.
In vivo Bioequivalence Studies
The following sequence of criteria is useful in assessing the need for in-vivo studies:
- Oral immediate release products with systemic action
- Indicated for serious conditions requiring assured response (Like aspirin 300mg)
- Narrow therapeutic margin.
- Drugs whose absorption is < 70%
- Unfavourable physiochemical properties, e.g. low solubility (<5mg/mL), low dissolution rate.
- Documented evidence for bioavailability problems.
- Non-oral immediate release products.
- Modified release products with systemic action.
In vivo bioequivalence studies are conducted in the usual manner as discussed forbioavailability studies, i.e. the pharmacokinetic and the pharmacodynamic methods.
In vitro Bioequivalence Studies
If none of the above criteria are applicable, comparative in-vitro dissolution studies will be sufficient. In vitro studies, i.e. dissolution studies can be used instead of in-vivo bioequivalence under certain circumstances, called as biowaivers (exemptions) –
- The drug product differs only in strength of the active substance it contains, provided all the following conditions hold –
- The qualitative composition is the same
- The ratio between active substance and the excipients is the same, or (in the case of small strengths) the ratio between the excipients is the same.
- Both products are produced by the same manufacturer at the same production site.
- Under the same test conditions, the in vitro dissolution rate is the same.
- The drug product has been slightly reformulated or the manufacturing method has been slightly modified by the original manufacturer in ways that can convincingly be argued to be irrelevant for the bioavailability (change in color, flavor and preservatives).
- The product contains no excipients known to significantly affect absorption of the active ingredient.
Moreover, - The product is intended for topical administration (cream, ointment, gel, etc.) for local effects.
- The product is for oral administration but not intended to be absorbed (antacid or radio-opaque medium).
- The product is administered by inhalation as a gas or vapors.
Bioequivalence Experimental Study Design
The various types of test designs that are usually employed in clinical trials, bioavailability and bioequivalence studies are discussed below.
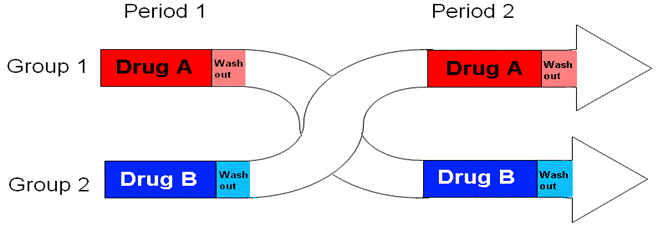
1. The Standard: Randomized, 2×2 Crossover Design
The most common and globally accepted method for conducting bioequivalence study is the Randomized, Two-Period, Two-Sequence, Single-Dose Crossover Design.
Here is how it works in practice:
- Subjects: A group of healthy human volunteers (usually 24 to 36 people) is selected.
- Group Division: They are randomly divided into two equal groups (Sequence 1 and Sequence 2).
- Period I:
- Group 1 receives the Test drug (the new generic).
- Group 2 receives the Reference drug (the innovator brand).
- Blood samples are collected at specific time intervals to measure drug concentration.
- The Washout Period: A gap of time where no drug is given. This ensures the first drug is completely eliminated from the body. Exam Key: The washout period must be at least 5 times the half-life (t1/2) of the drug.
- Period II (The “Crossover”):
- Group 1 now receives the Reference drug (the innovator brand).
- Group 2 now receives the Test drug (the new generic).
- Blood samples are collected again.
Why use a Crossover Design? Because every subject acts as their own “control.” This eliminates variations caused by different body weights, metabolisms, or genetics between different people.
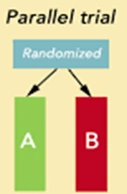
2. Parallel Design
In Parallel Study Design, subjects are randomized to receive one and only one treatment for the entire duration of the study. Unlike the crossover design, subjects do not switch treatments, and there is no washout period.
- Group A: Receives only the Test product (the new generic).
- Group B: Receives only the Reference product (the innovator brand).
The pharmacokinetic parameters (AUC, Cmax) are then calculated and compared statistically between the two separate groups.
When is Parallel Design Used?
- Drugs with Extremely Long Half-Lives (t1/2).
- Depot Formulations / Implants:
- Injectables designed to release medication slowly over 3 to 6 months cannot be easily studied in a crossover format because the drug remains in the system for an extensive period.
- High Toxicity Drugs:
- For highly toxic drugs (like specific chemotherapeutic agents), exposing a healthy volunteer or even a patient to multiple doses of different formulations (as required in a crossover) is unethical.
- Curative Treatments:
- If a drug cures a condition, the patient’s baseline health changes after the first period. You cannot cross them over to a second treatment because they are no longer in the same physiological state
3. Latin Square Design
What is a Latin Square Design?
A Latin square design is particularly used when there are more than two treatments. An experiment involving “n” treatments, than “n2” experimental units. A Latin Square Design is an experimental layout used to control two distinct sources of nuisance variability (background noise) simultaneously. It is called a “square” because the number of treatments being tested must be exactly equal to the number of rows and columns in the design.
- The Golden Rule: Just like a Sudoku puzzle, every treatment (usually represented by letters like A, B, C) must appear exactly once in every row and exactly once in every column.
Application in Clinical & Bioequivalence Studies
In a standard 2×2 crossover study, you compare 2 drugs over 2 periods. But what if you need to compare 3 different formulations (e.g., an Innovator brand, Generic A, and Generic B)?
Using a 3×3 Latin Square Crossover Design solves this perfectly:
- Rows represent: Subject Groups (or Sequences).
- Columns represent: Dosing Periods (with washout phases in between).
- Letters represent: The Treatments (A, B, and C).
Example of a 3×3 Latin Square:
- Sequence 1: Period 1 (Drug A/reference) → Period 2 (Drug B) → Period 3 (Drug C)
- Sequence 2: Period 1 (Drug B) → Period 2 (Drug C) → Period 3 (Drug A/reference)
- Sequence 3: Period 1 (Drug C) → Period 2 (Drug A/reference) → Period 3 (Drug B)
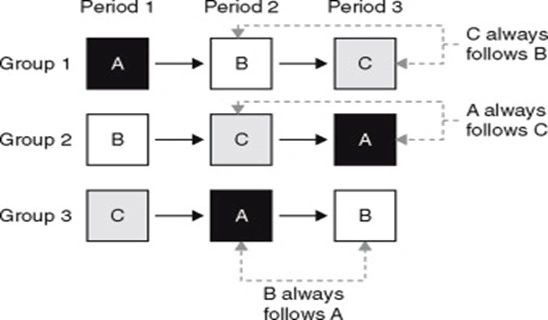
Notice how in Period 1, all three drugs are being tested simultaneously across different groups, and by the end of the study, every group has taken every drug.
Statistical Interpretation of Bioequivalence Data (Same for all 3 designs discussed above)
After the data has been collected, statistical methods must be applied to determine the level of significance of any observed difference in the rate and/or extent of absorption in order to establish bioequivalence between two or more drug products. The commonly adopted approaches to determine statistical differences are –
1. Analysis of variance (ANOVA) is a statistical procedure used to test the data fordifferences within and between treatment and control groups. A statistical difference between the pharmacokinetic parameters obtained from two or more drug products is considered statistically significant if there is a probability of less than 1 in 20 or 0.05 (p ≤ 0.05). The probability p is used to indicate the level of statistical significance. If p ≥ 0.05, the differences between the two drug products are not considered statistically significant.
2. Statistical Acceptance Criteria (The “80-125 Rule”): This is the most critical regulatory standard for bioequivalence (mandated by CDSCO, US FDA, and EMA).
For the generic drug to pass, researchers calculate the 90% Confidence Interval of the ratio between the Test and Reference drug for both AUC and Cmax.
The Rule: The generic drug’s parameters must fall strictly within the 80% to 125% limit of the reference drug.
Methods to enhance the dissolution rates and bioavailability of poorly soluble drugs.
There are several ways by which drug solubility or the dissolution rate can be enhanced.
Some of the widely used methods are discussed briefly.
- Micronization
- Nanonisation
- Supercritical fluid recrystallization
- Use of surfactants
- Use of salt forms
- Use of precipitation inhibitors
- Use of Amorphs, Anhydrates, Solvents and Metastable Polymorphs
- Solvent Depositions
- Precipitation
- Selective Adsorption on Insoluble Careers
- Solid Dispersions
- Molecular Encapsulation with cyclodextrins
- Micronization: The process involves reducing the size of the solid drug particlesto 1 to 10 microns commonly by spray drying or by use of air attrition methods (fluid energy or jet mill). The process is also called as micro-milling. Examples of drugs whose bioavailability have been increased by micronization include griseofulvin and several steroidal and sulpha drugs.
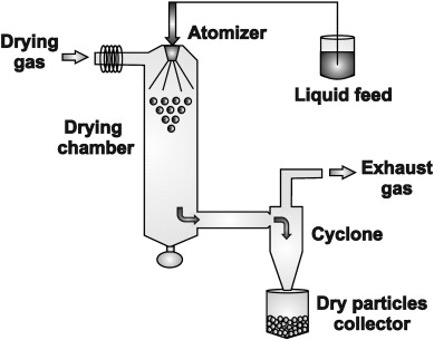
- Nanonisation: It‘s a process whereby the drug powder is converted tonanocrystals of sizes 200 – 600 nm, e.g. amphotericin B. The main production technologies currently in use to produce drug nanocrystals yield as a product a dispersion of drug nanocrystals in a liquid, typically water (called nanosuspension). There are three basic technologies currently in use to prepare nanoparticles:
- Pearl milling
- Homogenisation in water (wet milling as in a colloid mill)
- Homogenisation in non-aqueous media or in water with water-miscible liquids.
- Supercritical Fluid Recrystallization: It is a process in which a supercritical fluid (commonly carbon dioxide) is used to dissolve a solute and then recrystallize it under controlled temperature and pressure conditions to obtain purified crystals with desired particle size.
Carbon Dioxide (CO2) is the most widely used SCF in pharmaceuticals because it is non-toxic, non-flammable, inexpensive, and has a relatively low critical temperature (31.1°C), which protects heat-sensitive drugs. - Use of Surfactants: Surfactants are very useful as absorption enhancers andenhance both dissolution rate as well as permeability of drug. They enhance dissolution rate primarily by promoting wetting and penetration of dissolution fluid into the solid drug particles.
Nonionic surfactants like polysorbates are widely used. Examples of drugs whose bioavailability have been increased by use of surfactants in the formulation include steroids like spironolactone. - Use of Salt Forms: Salt formation is a commonly used method to enhance solubility of poorly soluble drugs by converting weak acids or bases into their salt forms. Weak acidic drugs form salts with bases, while weak basic drugs form salts with acids. Salt forms increase ionization, improve wettability, and enhance dissolution rate, leading to improved bioavailability. Examples include diclofenac sodium, morphine hydrochloride, and atropine sulfate. Although salt formation improves solubility, it may cause hygroscopicity and stability issues in some cases.
- Use of Precipitation Inhibitors: A significant increase in free drug concentrationabove equilibrium solubility results in supersaturation, which can lead to drug precipitation or crystallization. This can be prevented by use of inert polymers such HPMC, PVP, PVA, PEG, etc. which act by one or more of the following mechanisms –
- Increase the viscosity of crystallization medium thereby reducing the crystallization rate of drugs.
- Provide a steric barrier to drug molecules and inhibit crystallization through specific intermolecular interactions on growing crystal surfaces.
- Use of Amorphs, Anhydrates, Solvates and Metastable Polymorphs: Depending upon the internal structure of the solid drug, selection of proper form of drug with greater solubility is important. In general, amorphs are more soluble than metastable polymorphs, anhydrates are more soluble than hydrates and solvates are more soluble than non-solvates.
- Amorphous > Metastable > Stable.
- Solvent Deposition: Solvent deposition is a technique used to enhance the dissolution rate and bioavailability of poorly soluble drugs by dissolving the drug in a solvent and depositing it onto an inert carrier, followed by removal of the solvent.
This results in the drug being uniformly distributed in fine form over the surface of the carrier, which improves dissolution.
In this method, the poorly aqueous soluble drug such asnifedipine is dissolved in an organic solvent like alcohol and deposited on an inert, hydrophilic, solid matrix such as starch or microcrystalline cellulose by evaporation of solvent. - Precipitation: In this method, the poorly aqueous soluble drug such ascyclosporine is dissolved in a suitable organic solvent followed by its rapid mixing with a non-solvent to effect precipitation of drug in nanosize particles. The product so prepared is also called as hydrosol.
- Selective Adsorption on Insoluble Carriers: A highly active adsorbent such asthe inorganic clays like bentonite can enhance the dissolution rate of poorly water-soluble drugs such as griseofulvin, indomethacin and prednisone by maintaining the concentration gradient at its maximum. The two reasons suggested for the rapid release of drugs from the surface of clays are—the weak physical bonding between the adsorbate and the adsorbent, and hydration and swelling of the clay in the aqueous media.
- Solid Dispersions: These are generally prepared by solvent or co-precipitation method whereby both the guest solute and the solid carrier solvent are dissolved in a common volatile liquid solvent such as alcohol. The liquid solvent is removed by evaporation under reduced pressure or by freeze-drying which results in amorphous precipitation of guest in a crystalline carrier. e.g.amorphous sulphathiazole in crystalline urea. Such dispersions are often called as co-evaporates or co-precipitates. The method is suitable for thermolabile substances but has a number of disadvantages like higher cost of processing, use of large quantities of solvent, difficulty in complete removal of solvent, etc. With glassy materials, the dispersions formed are called as glass dispersions or glass suspensions. Fig. 11.9 shows comparative dissolution rates ofgriseofulvin from PVP dispersions. Other polymers such as PEG and HPMC are also employed to prepare solid dispersions of poorly water-soluble drugs such as nifedipine and itraconazole.
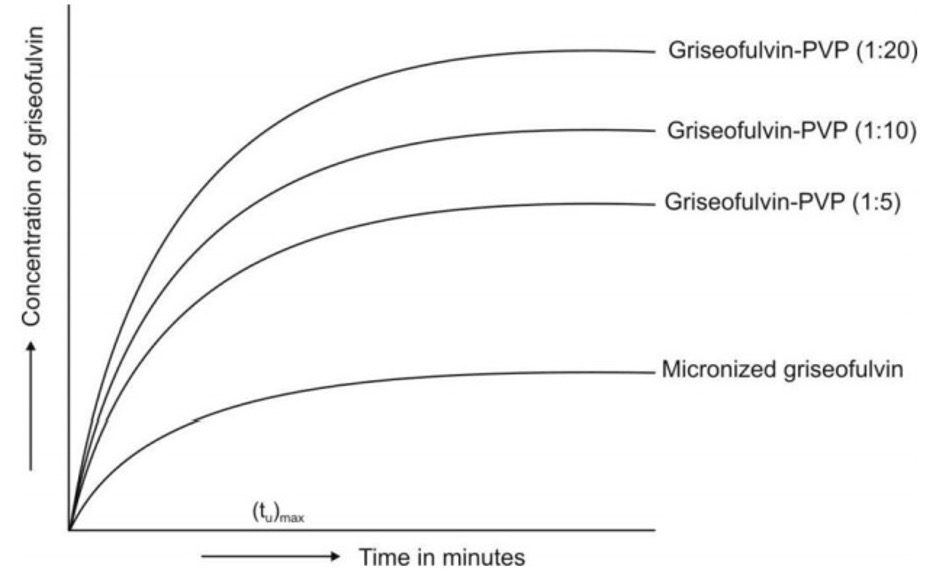
- Molecular Encapsulation with Cyclodextrins: The beta- and gamma-cyclodextrins and several of their derivatives are unique in having the ability to form molecular inclusion complexes with hydrophobic drugs having poor aqueous solubility.These bucket-shaped oligosaccharides produced from starch are versatile in having a hydrophobic cavity of size suitable enough to accommodate the hydrophobic/lipophilic drugs as guests; the outside of the host molecule is relatively hydrophilic (Fig. 11.10). Thus, the molecularly encapsulated drug has greatly improved aqueous solubility and dissolution rate. There are several examples of drugs with improved bioavailability due to such a phenomenon — thiazide diuretics, barbiturates, benzodiazepines and a number of NSAIDs.
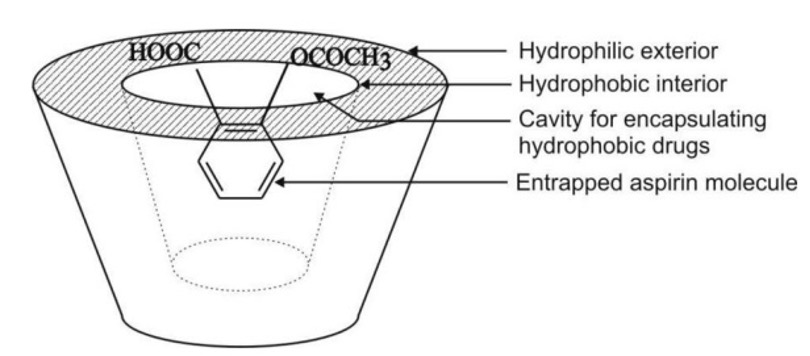
Questions:
10 marks.
- Define bioavailability. Explain any two methods for measurement of bioavailability.
- Write in detail about various approaches for improving dissolution of poorly soluble drugs.
- Discuss in detail drug metabolism and metabolism pathways of renal excretion.
- Define bioavailability. Explain pharmacokinetic method of assessment of bioavailability.
- Define metabolism, explain phase I reactions.
- Define and classify bioavailability, write about bioequivalence study.
5 marks.
- Define clearance. Write a note on renal clearance.
Or
How will you determine renal clearance. - Define dissolution. Explain various methods to enhance the dissolution of poorly soluble drugs.
- Explain about bioequivalence studies.
- Write the advantages and limitations of multiple drug or dose study.
- Write a note of determination (measurement) of bioavailability.
- In-vitro in-vivo correlation.
- Discuss the various study designs for performing bioequivalence studies.
- Explain the non-renal route of drug excretion.
2 marks.
- Define IVIVC
- Enumerate the different methods to enhance the dissolution of poorly soluble drugs.
- Objectives of bioavailability.
- Write limitations of urinary exception data for calculation of pharmacokinetics.
- List out non-renal route of drug excretion.
- Define absolute and relative bioavailability.
- What is clearance? Give the formula for same.
- Give the significance of bioequivalence.
- Write the formula to calculate hepatic excretion ratio.