
Unit-I: Introduction to Biopharmaceutics, Absorption and Distribution (10 Hrs.)
Syllabus:
Introduction to Biopharmaceutics.
Absorption: Mechanisms of drug absorption through GIT, factors influencing drug absorption though GIT, absorption of drug from Non per oral extra-vascular routes,
Distribution: Tissue permeability of drugs, binding of drugs, apparent, volume of drug distribution, plasma and tissue protein binding of drugs, factors affecting protein-drug binding. Kinetics of protein binding, Clinical significance of protein binding of drugs.
Introduction to Biopharmaceutics:
- Biopharmaceutics is a combination of two words i.e., Bio and Pharmaceutics.
Bio– Living organism/ living thing/ Living being.
Pharmaceutics– is the branch of pharmaceutical science that deals with the formulation, preparation, manufacture, and dispensing of drugs into suitable dosage forms. Means it deals with drug, dose, dosage form and dosage regimen. - Pharmacokinetics is a combination of two words i.e., Pharmacon and Kinetics.
Pharmacon– Drug
Kinetics– Movement/ Motion - Drugs, whether obtained from plant, animal or mineral sources or synthesized chemically, are rarely administered in their pure chemical form. Often, they are combined with a number of inert substances (excipients/adjuvants) and transformed into a convenient dosage form that can be administered by a suitable route.
- Earlier, it was believed that the therapeutic response by the body to a drug is an characteristic of its intrinsic pharmacological activity of the drug. But today, it is very much understood that the dose-response relationship obtained after drug administration by different routes- for example, oral and parenteral, are not the same.
- Variations are also observed when the same drug is administered as different dosage forms or similar dosage forms produced by different manufacturers, which in turn depend upon the physicochemical properties of the drug, the excipients present in the dosage form, the method of formulation and the manner of administration (dosage form). A new and separate discipline called biopharmaceutics has therefore been developed to account for all such factors that influence the therapeutic effectiveness of a drug.
- Biopharmaceutics is defined as the study of factors influencing the rate and amount of drug that reaches the systemic circulation and the use of this information to optimize the therapeutic efficacy of the drug products.
- Drug absorption is defined as the process of movement of unchanged drug from the site of administration to systemic circulation.
- Hence, Biopharmaceutics can also be defined as the study of factors influencing the rate and amount of drug absorption.
- Bioavailability is defined as the rate and extent (amount) of drug absorption.
- Any alteration in the drug’s bioavailability is reflected in its pharmacological effects.
- With bioavailability, there are other processes that also play a role in the therapeutic activity of a drug, they are distribution, elimination (metabolism and excretion).
- Drug Distribution is the movement of drug between one compartment and the other (generally blood and the extravascular tissues).
- Elimination is defined as the process that tends to remove the drug from the body and terminate its action.
- Elimination occurs by two processes—biotransformation (metabolism),which usually inactivates the drug, and excretion which is responsible for the exit of drug/metabolites from the body.
- In order to administer drugs optimally, knowledge is needed not only of the mechanisms of drug absorption, distribution, metabolism and excretion (ADME) but also of the rate (kinetics) at which they occur i.e. pharmacokinetics.
- Pharmacokinetics is defined as the study of time course of drug ADME and their relationship with its therapeutic and toxic effects of the drug.
- The use of pharmacokinetic principles in optimizing the drug dosage to suit individual patient needs and achieving maximum therapeutic utility is called as clinical pharmacokinetics.
- Below Figure is a schematic representation of processes comprising the pharmacokinetics of a drug.
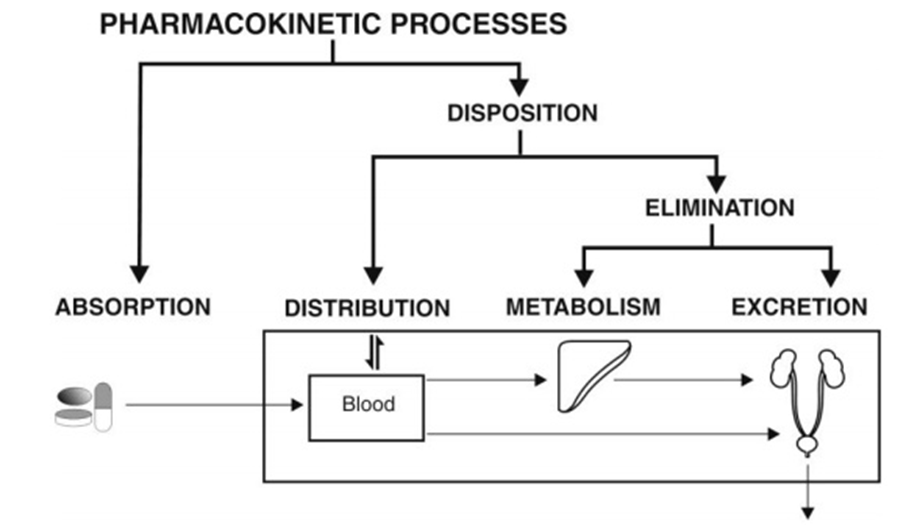
Fig. Schematic illustration of pharmacokinetic processes
Drug administration and therapy can now be conveniently divided into four phases or processes:
- The Pharmaceutical Phase: It is concerned with–
(a) Physicochemical properties of the drug, and
(b) Design and manufacture of an effective drug product for administration by a suitable route. - The Pharmacokinetic Phase: It is concerned with the ADME of drugs as elicited by the plasma drug concentration-time profile and its relationship with the dose, dosage form and frequency and route of administration. In short, it is the sum of all the processes inflicted by the body on the drug.
- The Pharmacodynamic Phase: It is concerned with the biochemical and physiological effects of the drug and its mechanism of action.
Thus, in comparison –
Pharmacokinetics is a study of what the body does to the drug, whereas,
Pharmacodynamics is a study of what the drug does to the body.
Pharmacokinetics relates changes in concentration of drug within the body with time after its administration, whereas
Pharmacodynamics relates response to concentration of drug in the body. - The Therapeutic Phase: It is concerned with the translation of pharmacological effect into clinical benefit.
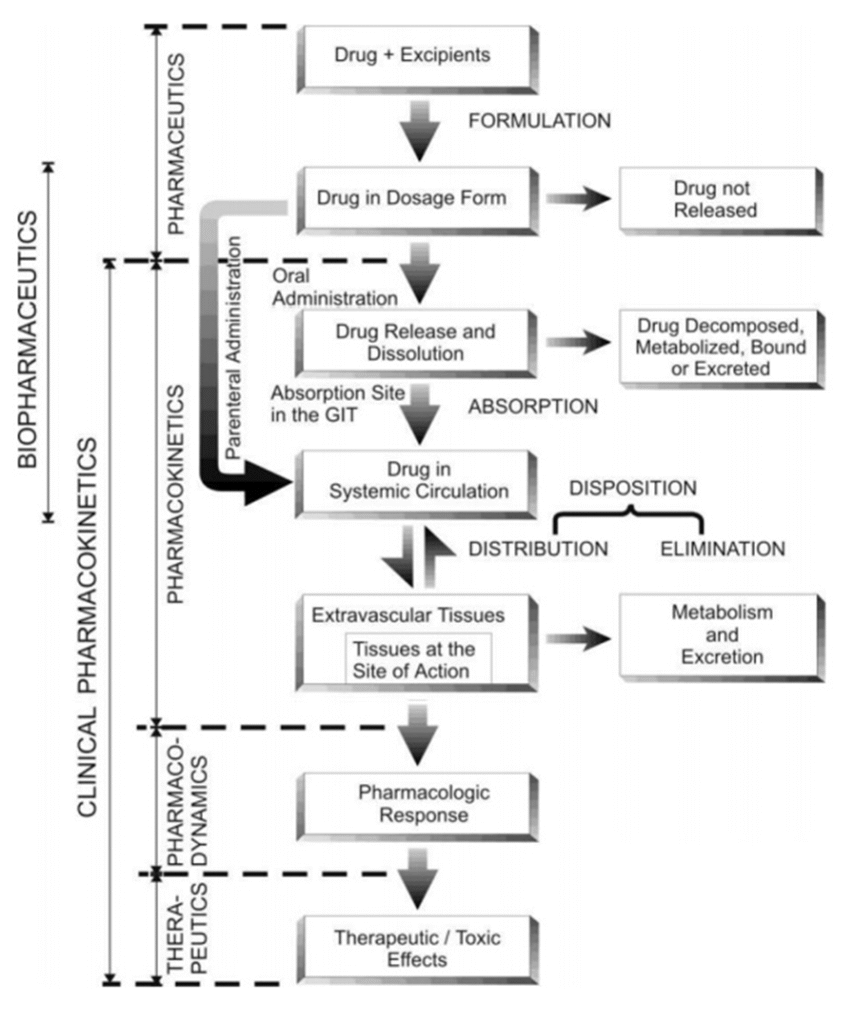
Fig. Schematic representation of the processes involved in drug therapeutics
- To achieve optimal therapy with a drug, the drug product must be designed to deliver the active principle at an optimal rate and amount, depending upon the patient’s needs. Knowledge of the factors affecting the bioavailability of drug helps in designing such an optimum formulation and saves many drugs that may be discarded as useless. On the other hand, rational use of the drug or the therapeutic objective can only be achieved through a better understanding of pharmacokinetics (in addition to pharmacodynamics of the drug), which helps in designing a proper dosage regimen (the manner in which the drug should be taken).
- The knowledge and concepts of biopharmaceutics and pharmacokinetics thus have an integral role in the design and development of new drugs and their dosage forms and improvement of therapeutic efficacy of existing drugs.
Absorption of Drugs:
- A drug injected intravascularly (intravenously and/or intra-arterially) directly enters the systemic circulation and exerts its pharmacological effects.
- However, majority of drugs are administered extra-vascularly, generally orally. If intended to act systemically, such drugs can exert their pharmacological actions only when they come into blood circulation from their site of application, and for this, absorption is an important prerequisite step.
- Drug absorption is defined as the process of movement of unchanged drug from the site of administration to systemic circulation.
- Followingabsorption, the effectiveness of a drug can only be assessed by its concentration at the site of action. But, it is difficult to measure the drug concentration at such a site. Instead, the concentration can be measured more accurately in plasma. There always exist a correlation between the plasma concentration of a drug and the therapeutic response and thus, absorption can also be defined as the process of movement of unchanged drug from the site of administration to the site of measurement i.e. plasma. This definition takesinto account the loss of drug that occurs after oral administration due to pre-systemic metabolism or first-pass effect.
- Not only the magnitude of drug that comes into the systemic circulation but also the rate at which it is absorbed is important. This is clear from Below Fig.
- A drug that is completely but slowly absorbed may fail to show therapeutic response as the plasma concentration for desired effect is never achieved. On the contrary, a rapidly absorbed drug attains the therapeutic level easily to elicit pharmacological effect.
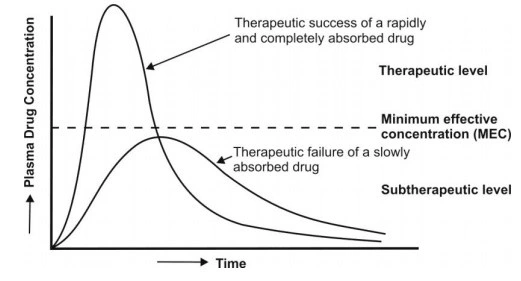
Fig. Plots showing significance of rate and extent of absorption in drug therapy.
- Thus, both the rate and the extent of drug absorption are important. Such an absorption pattern has several advantages:
1. Lesser susceptibility of the drug for degradation or interaction due to rapid absorption.
2. Higher blood levels and rapid onset of action.
3. More uniform, greater and reproducible therapeutic response. - Drugs that have to enter the systemic circulation to exert their effect can be administered by three major routes:
1. The Enteral Route: includes peroral i.e. gastrointestinal, sublingual/buccal and rectal routes. The GI route is the most common for administration of majority of drugs.
2. The Parenteral Route: includes all routes of administration through or under one or more layers of skin. While no absorption is required when the drug is administered I.V., it is necessary for extravascular parenteral routes like the subcutaneous and the intramuscular routes.
3. The Topical Route: includes skin, eyes or other specific membranes. The intranasal, inhalation, intravaginal and transdermal routes may be considered enteral or topical according to different definitions.
Mechanisms of drug absorption through GIT:
Gastrointestinal Absorption of Drugs
- The oral route of drug administration is the most common for systemically acting drugs and therefore, more importance will be given to gastrointestinal (GI) absorption of drugs.
- Among all dosage forms, about 80% are oral dosage forms (70% tablets and 10% other orals) and remaining 20% are other dosage forms.
- Moreover, it covers all the aspects (or factors) of variability observed in drug absorption. Before proceeding to discuss absorption aspects, a brief description of cell membrane structure and physiology is necessary.
Cell Membrane: Structure and Physiology
- For a drug to be absorbed and distributed into organs and tissues and eliminated from the body, it must pass through one or more biological membranes/barriers at various locations. Such a movement of drug across the membrane is called asdrug transport.
- The basic structure of cell membrane is shown in below Fig.
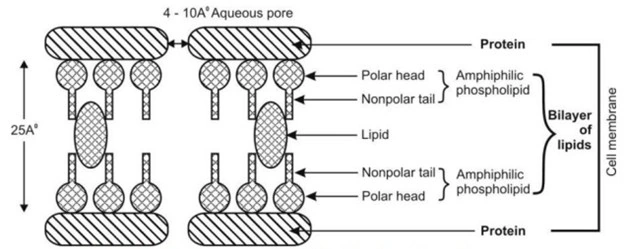
- The cellular membrane consists of a double layer of amphiphilic phospholipid molecules arranged in such a fashion that their hydrocarbon chains are oriented inwards to form the hydrophobic or lipophilic phase and their polar heads oriented to form the outer and inner hydrophilic boundaries of the cellular membrane that face the surrounding aqueous environment. Globular protein molecules are associated on either side of these hydrophilic boundaries and also combined within the membrane structure. In short, the membrane is a mayonnaise sandwich where a bimolecular layer of lipids is contained between two parallel monomolecular layers of proteins.
- The hydrophobic core of the membrane is responsible for the relative protection of polar molecules.
- Aqueous filled pores or perforations of 4 to 10 Å in diameter are also present in the membrane structure through which inorganic ions and small organic water-soluble molecules like urea can pass. In general, the bio-membrane acts like a semipermeable barrier permitting rapid and limited passage of some compounds while restricting that of others.
- The GI lining constituting the absorption barrier allows most nutrients like glucose, amino acids, fatty acids, vitamins, etc. to pass rapidly through it into the systemic circulation but prevents the entry of certain toxins and medicaments. Thus, for a drug to get absorbed after oral administration, it must first pass through this biological barrier.
Mechanisms of Drug Absorption:
The three broad categories of drug transport mechanisms involved in absorption are –
A. Transcellular/intracellular transport
B. Paracellular/intercellular transport
C. Vesicular transport
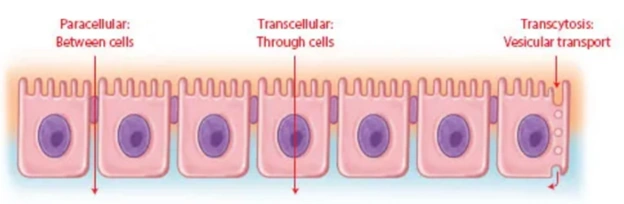
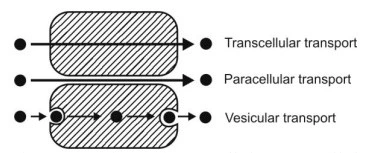
Fig. compares transcellular, paracellular and vesicular transport mechanisms.
A. Transcellular/Intracellular Transport
Transcellular/Intracellular Transport –is defined as the passage of drugs across the GI epithelium. It is the most common pathway for drug transport.
The various transcellular transport processes involved in drug absorption are –
- Passive Transport Processes –These transport processes do not require energy other than that of molecular motion (Brownian motion) to pass through the lipid bilayer. Passive transport processes can be further classified into following types –
a. Passive diffusion.
b. Pore transport.
c. Ion-pair transport.
d. Facilitated- or mediated-diffusion. - Active Transport Processes –This transport process requires energy from ATP to move drug molecules from extracellular (outside the cell) to intracellular milieu (inside the cell membrane). These are of two types –
a. Primary active transport.
b. Secondary active transport – this process is further subdivided into two –
i. Symport (co-transport).
ii. Antiport (counter-transport).
B. Paracellular/Intercellular Transport
Paracellular/Intercellular Transport –is defined as the transport of drugs through the junctions between the GI epithelial cells. This pathway is of minor importance in drug absorption.
The two paracellular transport mechanisms involved in drug absorption are –
- Permeation through tight junctions of epithelial cells –this process basically occurs through openings which are little bigger than the aqueous pores. Compounds such as insulin and cardiac glycosides are taken up this mechanism.
- Persorption –is permeation of drug through temporary openings formed by shedding of two neighboring epithelial cells into the lumen.
C. Vesicular or Corpuscular Transport (Endocytosis)
Vesicular or Corpuscular Transport (Endocytosis): Vesicular transport is the active process by which cells move large molecules or bulk materials using small, membrane-bound sacs called vesicles. Since the mechanism involves transport across the cell membrane, the process can also be classified as transcellular.
Vesicular transport of drugs can be classed into two categories –
- Pinocytosis.
- Phagocytosis
Mechanisms of Drug Absorption Explanation
Passive Diffusion/ Transport:
- Also called non-ionic diffusion, it is the major process for absorption of more than 90% of the drugs.
- The driving force for this process is the concentration or electrochemical gradient.
- It is defined as the difference in the drug concentration on either side of the membrane.
- Drug movement is a result ofthe kinetic energy of molecules.
- Since no energy source is required, the process is called as passive diffusion.
- During passive diffusion, the drug present in the aqueous solution at the absorption site (GIT) partitions and dissolves in the lipid material of the cell membrane and finally leaves it by dissolving again in an aqueous medium, this time at the inside of the membrane.
- Passive diffusion is best expressed by Fick’s first law of diffusion, which states that the drug molecules diffuse from a region of higher concentration to one of lower concentration until equilibrium is attained and that the rate of diffusion is directly proportional to the concentration gradient across the membrane. It can be mathematically expressed by the following equation:
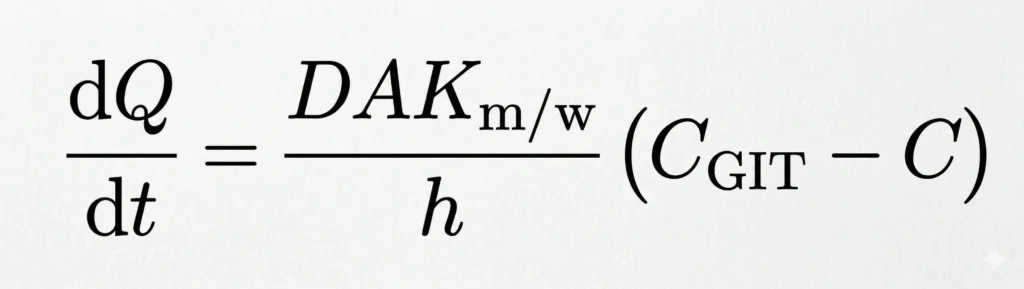
Where,
dQ/dt = rate of drug diffusion (amount/time). It also represents the rate of appearance of drug in blood
D = diffusion coefficient of the drug through the membrane (area/time)
A = surface area of the absorbing membrane for drug diffusion (area)
Km/w = partition coefficient of the drug between the lipoidal membrane and the aqueous GI fluids (no units)
(CGIT – C) = difference in the concentration of drug in the GI fluids and the plasma, called as the concentration gradient (amount/volume)
h = thickness of the membrane (length)
- Based on the above equation, certain characteristics of passive diffusion can be generalized-
- The drug moves down the concentration gradient, indicating downhill transport.
- The process is energy-independent and non-saturable.
- The rate of drug transfer is directly proportional to the concentration gradient between GI fluids and the blood compartment.
- Greater the area and lesser the thickness of the membrane, faster the diffusion; thus, more rapid is the rate of drug absorption from the intestine than from the stomach.
- The process is rapid over short distances and slower over long distances.
- Equilibrium is attained when the concentration on either side of the membrane becomes equal.
- Drugs which can exist in both ionized and unionized forms, the equilibrium achieved primarily by the transfer of the unionized species; because, the rate of transfer of unionized species is 3 to 4 times more than the rate for ionized drugs.
- Greater the membrane/water partition coefficient of drug, faster the absorption; since the membrane is lipoidal in nature, a lipophilic drug diffuses at a faster rate by solubilizing in the lipid layer of the membrane.
- The drug diffuses rapidly when the volume of GI fluid is low; conversely, dilution of GI fluids decreases the drug concentration in these fluids (CGIT) and lower the concentration gradient (CGIT – C). This phenomenon is, however, made use of in treating cases of oral overdose or poisoning.
- The process is dependent, to a lesser extent,– drugs having molecular weights between 100 to 400 Daltons are effectively absorbed passively. The diffusion generally decreases with increase in the molecular weight of the compound. However, there are exceptions—for example, cyclosporin A, a peptide of molecular weight 1200, is absorbed orally much better than any other peptide.
- Initially, when the drug is ingested, CGIT >> C and a large concentration gradient exists thereby acting as the driving force for absorption. As equilibrium approaches, the drug diffusion should stop and consequently a large fraction of drug may remain unabsorbed. But this is not the case; once the passively absorbed drug enters blood, it is rapidly swept away and distributed into a much larger volume of body fluids and hence, the concentration of drug at the absorption site, CGIT, is maintained greater than the concentration of drug in plasma. Such a condition is called as sink condition for drug absorption.
- Since under usual conditions of absorption, D, A, Km/w and h are constants, the term DAKm/w/h can be replaced by a combined constant P called as permeability coefficient. Permeability refers to the ease with which a drug can penetrate or diffuse through a membrane. Moreover, due to sink conditions, the concentration of drug in plasma C is very small in comparison to CGIT. As a result, Fick’s first law of diffusion equation may be simplified to:
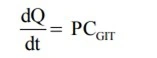
- Above Equation is an expression for a first-order process. Thus, passive diffusion follows first-order kinetics. Since a large concentration gradient always exists at the absorption site for passive diffusion, the rate of drug absorption is usually more rapid than the rate of elimination. Besides, dilution and distribution of the absorbed drug into a large pool of body fluids and its subsequent binding to various tissues are other reasons for elimination being slower than absorption.
- Below Figure illustrates the relative permeability of different molecules to lipid bilayer.
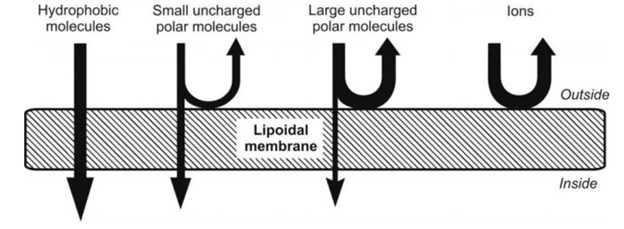
Fig. Relative passive diffusion rate of different types of molecules
Pore Transport:
- It is also called convective transport (Momentum within fluid), bulk flow or filtration. This mechanism is responsible for transport of molecules into the cell through the protein channels present in the cell membrane.
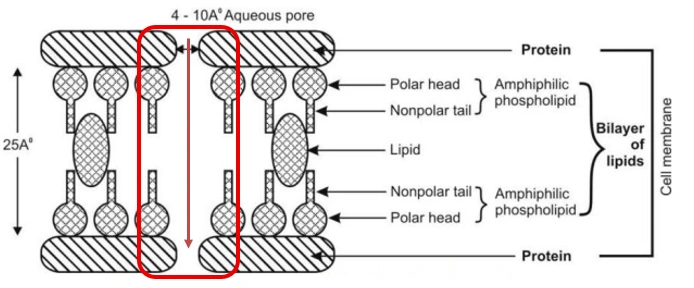
Fig. Pore transport illustration
- Following are the characteristics of pore transport –
- The driving force is constituted by the hydrostatic pressure or the osmotic differences across the membrane due to which bulk flow of water along with small solid molecules occurs through such aqueous channels. Water flux that promotes such a transport is called as solvent drag.
- The process is important in the absorption of low molecular weight (less than 100), low molecular size (smaller than the diameter of the pore) and generally water-soluble drugs like urea and sugars. flow through narrow, aqueous-filled channels or pores in the membrane structure.
- Chain-like or linear compounds of molecular weight up to 400 Daltons can be absorbed by filtration.
- Drug permeation through water-filled channels is of particular importance in renal excretion, removal of drug from the cerebrospinal fluid and entry of drugs into the liver.
Ion-Pair Transport:
- Yet another mechanism that explains the absorption of drugs like quaternary ammonium compounds(+) and sulphonic acids(-), which ionize under all pH conditions, is ion-pair transport.
- The charged drug molecules penetrate the membrane by forming reversible neutral complexes with endogenous ions (Within the body) of the GIT like mucin.
- Such neutral complexes have both the required lipophilicity as well as aqueous solubility for passive diffusion. Such a phenomenon is called as ion-pair transport (Fig. 2.5).
- Propranolol, a basic drug that forms an ion pair with oleic acid, is absorbed by this mechanism. Anti-cancer drugs and fat-soluble vitamins (A,D,E,K) sometimes absorb through this mechanism.
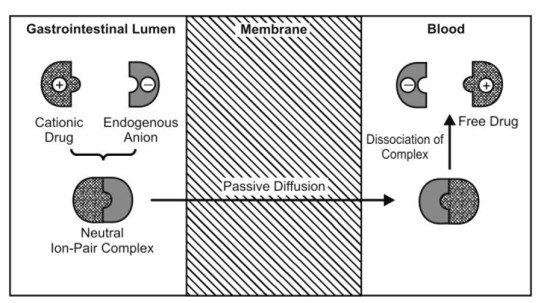
Fig. Ion pair transport illustration
Carrier-Mediated Transport:
- Some polar drugs (partial positive and negative charges) cross the membrane more readily than can be predicted from their concentration gradient and partition coefficient values.
- This suggests presence of specialized transport mechanisms without which many essential water-soluble nutrients like monosaccharides, amino acids and vitamins will be poorly absorbed.
- The mechanism involves a component of the membrane called as the carrier that binds reversibly or non-covalently with the solute molecules to be transported. This carrier-solute complex traverses across the membrane to the other side where it dissociates and discharges the solute molecule. The carrier then returns to its original site to complete the cycle by accepting a fresh molecule of solute.
- Carriers in membranes are proteins (transport proteins) and may be an enzyme or some other component of the membrane. They are numerous in all biological membranes and are found dissolved in the lipid bilayer of the membrane.
- Important characteristics of carrier-mediated transport are:
- A carrier protein always has an uncharged (non-polar) outer surface which allows it to be soluble within the lipid of the membrane.
- The carriers have no directionality; they work with same efficiency in both directions.
- The transport process is structure-specific i.e. the carriers have special affinity for and transfer a drug of specific chemical structure only (i.e. lock and key arrangement); generally, the carriers have special affinity for essential nutrients.
- As the number of carriers is limited, the transport system is subject to competition between agents having similar structure.
- Since the number of carriers is limited, the system is capacity-limited i.e. at higher drug concentration; the system becomes saturated and approaches an asymptote. It is important to note that for a drug absorbed by passive diffusion, the rate of absorption increases linearly with the concentration but in case of carrier-mediated processes, the drug absorption increases linearly with concentration until the carriers become saturated after which it becomes curvilinear and approach a constant value at higher doses (see Fig. 2.6). Such a capacity-limited process can be adequately described by mixed order kinetics, also called as Michaelis-Menten, saturation or non-linear kinetics. The process is called mixed-order because it is first-order at sub-saturation drug concentrations and apparently zero-order at and above saturation levels. Moreover, the capacity-limited characteristics of such a system suggest that the bioavailability of a drug absorbed by such a system decrease with increasing dose—for example, vitamins like B1, B2 and B12. Hence, administration of a large single oral dose of such vitamins is irrational.
- Specialized absorption or carrier-mediated absorption generally occurs from specific sites of the intestinal tract which are rich in number of carriers. Such an area in which the carrier system is most dense is called as absorption window. Drugs absorbed through such absorption windows are poor candidates for controlled release formulations.
- Comparison of rate of absorption versus drug concentration plots for passive and carrier-mediated transport processes.
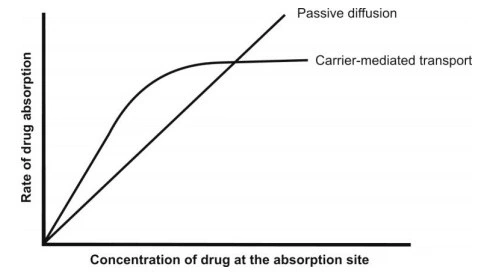
Fig. Comparison of rate of absorption versus drug concentration plots for passive and carrier-mediated transport processes
Facilitated Diffusion:
- It is a carrier-mediated transport system that operates down the concentration gradient (downhill transport) but at a much a faster rate than can be accounted by simple passive diffusion.
- The driving force is concentration gradient (hence a passive process). Since no energy expenditure is involved, the process is not inhibited by metabolic poisons that interfere with energy production.
- Facilitated diffusion is of limited importance in the absorption of drugs. Examples of such a transport system include entry of glucose into RBCs and intestinal absorption of vitamins B1 and B2. A classic example of passive facilitated diffusion is the GI absorption of vitamin B12.
- An intrinsic factor (IF), a glycoprotein produced by the gastric parietal cells, forms a complex with vitamin B12 which is then transported across the intestinal membrane by a carrier system.
- Facilitated diffusion of drug is illustrated in the below figure.
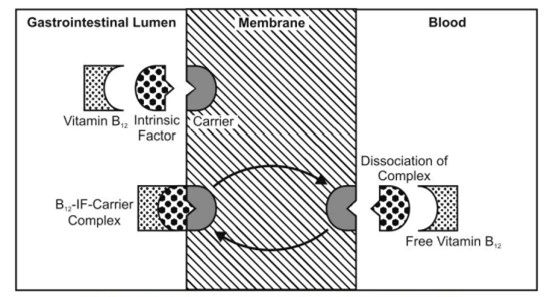
Fig. Facilitated diffusion illustration
Active Transport:
- This transport mechanism requires energy in the form of ATP.
- Active transport is responsible for transporting small foreign molecules (like drugs and toxins) especially out of cells which make them clinically important.
- Active transport is a more important process than facilitated diffusion in the absorption of nutrients and drugs and differs from it in several respects:
- The drug is transported from a region of lower concentration to higher concentration i.e. against the concentration gradient (in the case of ions, against an electrochemical gradient) or uphill transport, without any regard for equilibrium.
- The process is faster than passive diffusion.
- Since the process is uphill, energy is required in the work done by the carrier.
- As the process requires expenditure of energy, it can be inhibited by metabolic poisons that interfere with energy production like fluorides, cyanide and dinitrophenol and lack of oxygen, etc. Endogenous substances that are transported actively include sodium, potassium, calcium, iron, glucose, certain amino acids and vitamins like niacin, pyridoxin and ascorbic acid.
- Drugs having structural similarity to such agents are absorbed actively, particularly the agents useful in cancer chemotherapy. Examples include absorption of 5-fluorouracil and 5-bromouracil via the pyrimidine transport system, absorption of methyldopa and levodopa via an L-amino acid transport system and absorption of ACE inhibitor enalapril via the small peptide carrier system.
- Active transport is also important in renal and biliary excretion of many drugs and their metabolites and secretion of certain acids out of the CNS.
- Active transport of a drug is illustrated in the below figure.
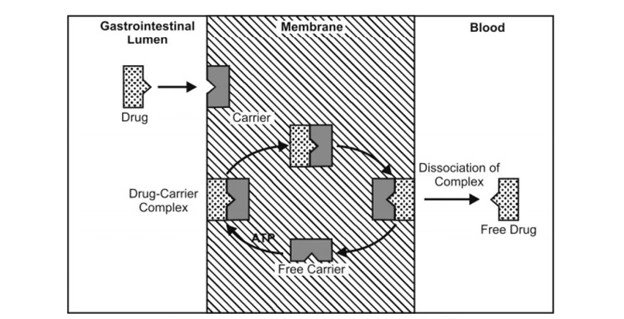
Fig. Active transport illustration
- Below Figure- compares active and passive transport
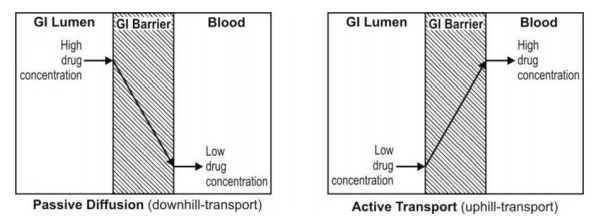
Fig. Comparison between active and passive transport
Comparison between Active Transport vs. Passive Transport
| Feature | Passive Diffusion | Active Transport |
| Driving Force | Concentration Gradient | Cellular Energy (ATP) |
| Direction | High to Low (Downhill) | Low to High (Uphill) |
| Carrier Protein | Not Required | Required |
| Energy (ATP) | Not Required | Required |
| Saturation | Not Saturable (Linear) | Saturable (Non-linear) |
| Selectivity | Non-specific | Highly Specific |
| Examples | Most drugs (Aspirin, Paracetamol) | Vitamins, Glucose, Levodopa, 5-Fluorouracil |
Endocytosis:
- It is a minor transport mechanism which involves engulfing extracellular materials within a segment of the cell membrane to form a saccule or a vesicle (hence also called as corpuscular or vesicular transport) which is then pinched-off intracellularly (see below Fig.). This is the only transport mechanism whereby a drug or compound does not have to be in an aqueous solution in order to be absorbed.
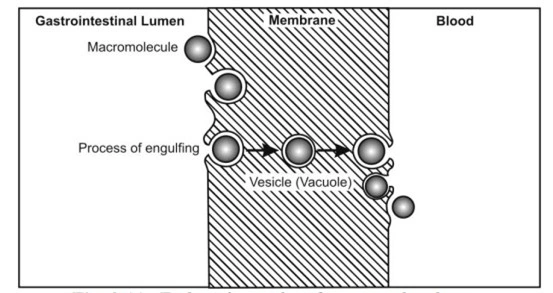
Fig. Endocytosis illustration
- This phenomenon is responsible for the cellular uptake of macromolecular nutrients like fats and starch, oil soluble vitamins like A, D, E and K, water soluble vitamin like B12 and drugs such as insulin.
- Another significance of such a process is that the drug is absorbed into the lymphatic circulation thereby bypassing first-pass hepatic metabolism.
- Endocytosis includes two types of processes:
- Pinocytosis (cell drinking): uptake of fluid solute, and
- Phagocytosis (cell eating): adsorptive uptake of solid particulates.
Combined Absorption Mechanisms:
- A drug might be absorbed by more than just one mechanism—for example, cardiac glycosides are absorbed both passively as well as by active transport. Vitamin B12 is absorbed by passive diffusion, facilitated diffusion as well as endocytosis. The transport mechanism also depends upon the site of drug administration.
- Absorption of drugs by various mechanisms is summarized in below Fig.
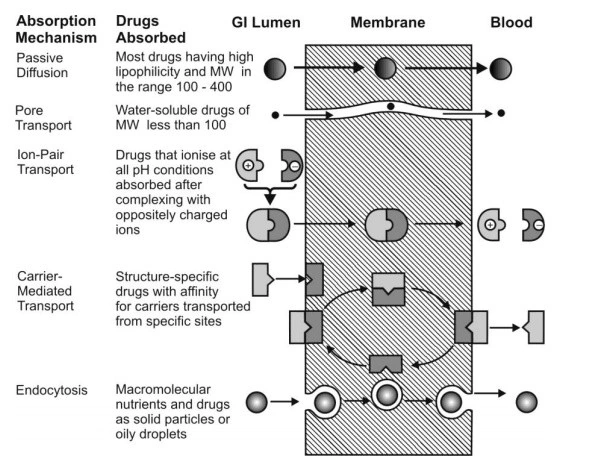
Fig. Summary of important transport processes and drugs absorbed through them
FACTORS INFLUENCING DRUG ABSORPTION AND BIOAVAILABILITY:
Biopharmaceutic Considerations in Dosage Form Design
- To achieve the desired therapeutic objective, the drug product must deliver the active drug at an optimal rate and amount.
- By proper biopharmaceutic design, the rate and extent of drug absorption can be varied from rapid and complete absorption to slow and sustained absorption depending upon the desired therapeutic objective. The chain of events that occur following administration of a solid dosage form such as a tablet or a capsule until its absorption into systemic circulation are depicted in below Fig.
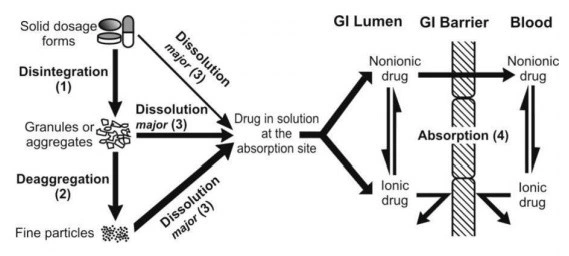
Fig. Sequence of events in the absorption of drugs from orally administered solid dosage forms
- The process consists of four steps:
- Disintegration of the drug product.
- Deaggregation and subsequent release of the drug.
- Dissolution of the drug in the aqueous fluids at the absorption site.
- Absorption i.e. movement of the dissolved drug through the GI membrane into the systemic circulation and away from the absorption site.
- As illustrated in above Fig., the drug may also dissolve before disintegration or deaggregation of the dosage form, and before or after reaching the absorption site. Unless the drug goes into solution, it cannot be absorbed into the systemic circulation.
- In a series of kinetic or rate processes, the rate at which the drug reaches the systemic circulation is determined by the slowest of the various steps involved in the sequence. Such a step is called as therate-determining orrate-limiting step (RDS). The rate and extent of drug absorption from itsdosage form can be influenced by a number of factors in all these steps. The various factors that influence drug absorption (also called as biopharmaceutic factors in the dosage form design) can be classified asshown below.
Factors influencing GI Absorption of a Drug from its Dosage Form:
A. PHARMACEUTICAL FACTORS:
Include factors relating to the physicochemical properties of the drug, and dosage form characteristics and pharmaceutical ingredients
I. Physicochemical Properties of Drug Substances (API):
- Drug solubility and dissolution rate
- Particle size and effective surface area
- Polymorphism and amorphism
- Pseudopolymorphism (hydrates/solvates)
- Salt form of the drug
- Lipophilicity of the drug
- pKa of the drug and gastrointestinal pH
- Drug stability
II. Dosage Form Characteristics and Pharmaceutical Ingredients (Pharmaco-technical Factors)
- Disintegration time (tablets/capsules)
- Dissolution time
- Manufacturing variables
- Pharmaceutical ingredients (excipients/adjuvants)
- Nature and type of dosage form
- Product age and storage conditions
B. BIOLOGICAL/ PATIENT RELATED FACTORS:
Include factors relating to the anatomical, physiological and pathological characteristics of the patient
- Age
- Gastric emptying time
- Intestinal transit time
- Gastrointestinal pH
- Disease states
- Blood flow through the GIT
- Gastrointestinal contents:
- Other drugs
- Food
- Fluids
- Other normal GI contents
- Presystemic metabolism by:
- Luminal enzymes
- Gut wall enzymes
- Hepatic enzymes
- Hepatic enzymes
Factors Explanation:
A. PHARMACEUTICAL FACTORS
PHYSICOCHEMICAL FACTORS AFFECTING DRUG ABSORPTION:
- Drug solubility & dissolution rate.
- Particle Size and Effective Surface Area of the Drug.
- Polymorphism and Amorphism
- Hydrates/Solvates (Pseudopolymorphism)
- Salt form of the drug
- Drug pKa and Lipophilicity and GI pH—pH Partition Hypothesis
- Drug stability
1. Drug Solubility and Dissolution Rate:
- An important prerequisite for the absorption of a drug by all mechanisms except endocytosis is that it must be present in aqueous solution. This in turn depends on the drug’s aqueous solubility and its dissolution rate.
- Absolute or intrinsic solubility is defined as the maximum amount of solute dissolved in a given solvent under standard conditions of temperature, pressure and pH. Itis a static property.
- Dissolution rate is defined as the amount of solid substance that goes into solution per unit time under standard conditions of temperature, pH and solvent composition and constant solid surface area. Itis a dynamic process.
- Except in case of controlled-release formulations, disintegration and deaggregation occur rapidly if it is a well-formulated dosage form. Thus, the two critical slower rate-determining processes in the absorption of orally administered drugs are:
- Rate of dissolution, and
- Rate of drug permeation through the bio-membrane.
- Dissolution is the RDS for hydrophobic, poorly aqueous soluble drugs like griseofulvin and spironolactone; absorption of such drugs is often said to be dissolution rate-limited.
- If the drug is hydrophilic with high aqueoussolubility—for example, cromolyn sodium or neomycin, then dissolution is rapid and RDS in the absorption of such drugs is rate of permeation through the bio-membrane. In other words, absorption of such drugs is said to be permeation rate-limited or transmembrane rate-limited.
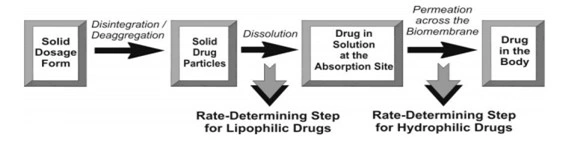
Fig. The two rate-determining steps in the absorption of drugs from orally administered formulations.
- Based on the intestinal permeability and solubility of drugs, Amidon et al developed Biopharmaceutics Classification System (BCS) which classifies the drugs into one of the 4 groups as shown in the below table.
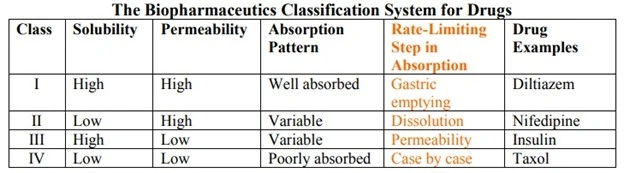
Class I drugs (high solubility/high permeability)are well absorbed orally since they have neither solubility nor permeability limitation.
Class II drugs (low solubility/high permeability)show variable absorption owing to solubility limitation.
Class III drugs (high solubility/low permeability)also show variable absorption owing to permeability limitation.
Class IV drugs (low solubility/low permeability)are poorly absorbed orally owing to both solubility and permeability limitations.
- Theories of Drug Dissolution
- Diffusion layer model/Film theory,
- Danckwert’s model/Penetration or Surface renewal theory, and
- Interfacial barrier model/Double-barrier or Limited solvation theory.
2. Particle Size and Effective Surface Area of the Drug:
- Particle size and surface area of a solid drug are inversely related to each other.
- Smaller the drug particle, greater the surface area. Two types of surface area of interest can be defined:
- Absolute surface area which is the total area of solid surface of any particle, and
- Effective surface area which is the area of solid surface exposed to the dissolution medium.
- Noyes-Whitney equation,
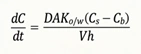
where,
D = diffusion coefficient (diffusivity) of the drug
A= surface area of the dissolving solid
Kw/o = water/oil partition coefficient of the drug considering the fact that dissolution body fluids are aqueous.
Since the rapidity with which a drug dissolves depends on the Kw/o, it is also called as the intrinsic dissolution rate constant. It is a characteristic of drugs.
V = volume of dissolution medium.
h= thickness of the stagnant layer.
(Cs – Cb) = concentration gradient for diffusion of drug.
- From the Noyes-Whitney equation, it is clear that larger the surface area, higher the dissolution rate. Since the surface area increases with decreasing particle size, a decrease in particle size, which can be accomplished by micronisation, will result in higher dissolution rates.
- However, it is important to note that it is not the absolute surface area but the effective surface area that is proportional to the dissolution rate.
- Greater the effective surface area, more intimate the contact between the solid surface and the aqueous solvent and faster the dissolution.
- But it is only when micronisation reduces the size of particles below 0.1 microns that there is an increase in the intrinsic solubility and dissolution rate of the drug. The surface of such small particles has energy higher than the bulk of the solid resulting in an increased interaction with the solvent. This is particularly true in case of drugs which are non-hydrophobic.
- Micronisation has in fact enabled the formulator to decrease the dose of certain drugs because of increased absorption efficiency—for example, the griseofulvin dose was reduced to half and that of spironolactone was decreased 20 times following micronisation.
- However, in case of hydrophobic drugs like aspirin, phenacetin and phenobarbital, micronisation actually results in a decrease in the effective surface area of such powders and thus, a fall in the dissolution rate. Three reasons have been suggested for such an outcome —
- The hydrophobic surface of the drug adsorbs air onto their surface which inhibit their wettability.
- The particles re-aggregate to form larger particles due to their high surface free energy, which either float on the surface or settle at the bottom of the dissolution medium.
- Electrically induced agglomeration owing to surface charges prevents intimate contact of the drug with the dissolution medium.
- The net result of these effects is that there is a decrease in the effective surface area available to the dissolution medium and therefore a fall in the dissolution rate.
- The absolute surface area of hydrophobic drugs can be converted to their effective surface area by:
- Use of surfactant as a wetting agent that –
Decreases the interfacial tension, and
Displaces the adsorbed air with the solvent.
For example, polysorbate 80 increases the bioavailability of phenacetin by promoting its wettability. - Adding hydrophilic diluents such as PEG, PVP, dextrose, etc. which coat the surface of hydrophobic drug particles and render them hydrophilic.
- Use of surfactant as a wetting agent that –
- Particle size reduction and subsequent increase in the surface area anddissolution rate is not advisable under following circumstances –
- When the drugs are unstable and degrade in solution form (penicillin G and erythromycin),
- When drugs produce undesirable effects (gastric irritation caused by nitrofurantoin)
- In addition to increasing the dissolution rate, the second mechanism by which a reduction in particle size improves drug dissolution is through an increase in its solubility. However, such an effect can only be achieved by reducing the particle size to a submicron level which is possible by use of one of the following specialized techniques such as formation of:
- Molecular dispersion/solid solution where the sparingly soluble drug is molecularly entrapped in the lattice (matrix) of a hydrophilic agent such as cyclodextrins.
- Solid dispersion where the drug is dispersed in a soluble carrier such as PVP, PEG, urea, etc.
3. Polymorphism and Amorphism
Depending upon the internal structure, a solid can exist either in a crystalline or amorphous form.
Polymorphism:
- When a substance exists in more than one crystalline form, the different forms are termed as polymorphs and the phenomenon as polymorphism.
- The polymorphs differ from each other with respect to their physical properties such as solubility, melting point, density, hardness and compression characteristics.
- Depending on their relative stability, one of the several polymorphic forms will be physically more stable than the others. Such a stable polymorph represents the lowest energy state, has highest melting point and least aqueous solubility.
- The remaining polymorphs are called as metastable forms which represent the higher energy state, have lower melting points and higher aqueous solubilities.
- Since the metastable forms have greater aqueous solubility, they show better bioavailability and are therefore preferred in formulations.
- For example, The polymorphic form III of riboflavin is 20 times more water-soluble than form I.
Amorphism:
- Some drugs can exist in amorphous form (i.e. having no internal crystal structure). Such drugs represent the highest energy state.
- They have greater aqueous solubility than the crystalline forms because the energy required to transfer a molecule is more for crystalline form and less for non-crystalline (amorphous) form — for example, the amorphous form of novobiocin is 10 times more soluble than the crystalline form.
- Thus, the order for dissolution of different solid forms of drugs is —
Amorphous > Metastable > Stable.
4. Hydrates/Solvates (Pseudopolymorphism)
- Pseudopolymorphism refers to the phenomenon where a substance forms different crystal structures—known as hydrates or solvates—due to the inclusion of solvent molecules within the crystal lattice.
- Where in true polymorphism, the chemical composition remains identical across different forms, but in pseudopolymorphs elemental compositions differ as there are added solvents.
- Hydrates: Crystal forms where water is the solvent incorporated into the lattice in a specific ratio. They are highly relevant in pharmaceuticals as water is safe for consumption.
- Solvates: Crystal forms where a solvent other than water (e.g., ethanol, methanol, or acetone) is trapped in the structure. These are often less common in final drug products due to potential solvent toxicity.
- Generally, the anhydrous form of a drug has greater aqueous solubility than the hydrates. This is because the hydrates are already in interaction with water and therefore have less space for further interaction with water in comparison to the anhydrates.
- For Example, the anhydrous form of theophylline and ampicillin have higher aqueous solubilities, dissolve at a faster rate and show better bioavailability in comparison to their monohydrate and trihydrate forms respectively.
- On the other hand, the organic (nonaqueous) solvates have greater aqueous solubility than the non-solvates—for example, the chloroform solvate of griseofulvin is more water-soluble than their Hydrate form.
- Like polymorphs, the solvates too differ from each other in terms of their physical properties. In case of organic solvates, if the solvent is toxic, they are not of therapeutic use.
5. Salt Form of the Drug
- Most drugs are either weak acids or weak bases. One of the easiest approaches to enhance the solubility and dissolution rate of such drugs is to convert them into their salt forms.
- Generally, with weakly acidic drugs, a strong base salt is prepared such as the sodium and potassium salts of barbiturates and sulphonamides.
- In case of weakly basic drugs, a strong acid salt is prepared like the hydrochloride or sulphate salts of several alkaloidal drugs.
- At a given pH of a bulk solution in GIT, the solubility of a drug, whether acidic/basic or its salt form is a constant. The influence of salt formation on the drug solubility, rate of dissolution and absorption can be explained by considering the pH of the diffusion layer and not the pH of the bulk of the solution.
- Owing to the increased pH of the diffusion layer (towards basic), the solubility and dissolution rate of a weak acid in this layer is promoted; and it is a known fact that higher pH favours the dissolution of weak acids. Thus, if dissolution is faster, absorption is bound to be rapid.
- In case of salts of weak bases, the pH of the diffusion layer will be lower (towards acidic) in comparison to that found with the free base form of the drug. Consequently, the solubility of a basic drug at this lower pH is enhanced.
6. Drug pKa and Lipophilicity and GI pH
- The pH partition theory (Brodie et al) explains in simple terms, the process ofdrug absorption from the GIT and its distribution across all biological membranes.
- The theory states that for drug compounds of molecular weight greater than 100 Dalton, which are primarily transported across the biomembrane by passive diffusion, the process of absorption is governed by:
- The dissociation constant (pKa) of the drug.
- The lipid solubility of the unionised drug (a function of drug Ko/w).
- The pH at the absorption site.
- Since most drugs are weak acids or weak bases, their degree of ionisation depends upon the pH of the biological fluid.
- And only the unionised fraction of drug, can permeate the membrane passively until the equilibrium is attained.
- The above statement of the hypothesis was based on the assumptions that:
- The GIT is a simple lipoidal barrier to the transport of drug.
- Larger the fraction of unionised drug, faster the absorption.
- Greater the lipophilicity (Ko/w) of the unionised drug, better the absorption.
7. Drug Stability
- Drug stability can be defined as the ability of the dosage form to maintain its chemical, physical, therapeutic and microbial integrity during its storage and until used by the patient.
- Factors affecting drug stability:
- Temperature: According to Arrhenius theory, increase in temperature leads to an increase in the rate of reaction, thus leads to degradation and formation of harmful metabolites resulting in a decrease in bioavailability.
- pH: Change in pH results in a change in the rate of decomposition of drugs. Most of the drugs are stable at a particular pH only (4-8 pH).
- Moisture: Water acts as a catalyst for many chemical reactions such as oxidation and hydrolysis. It promotes the growth of microorganisms.
- Incompatibilities: An interaction between the excipient and drug in a dosage form may sometime lead to the instability of the product.
- Many other factors like light, oxygen, type of packaging and type of dosage form may also affect drug stability.
DOSAGE FORM (PHARMACO-TECHNICAL) FACTORS
- Disintegration Time
- Manufacturing/Processing Variables
- Pharmaceutical Ingredients/Excipients (Formulation factors)
- Nature and Type of Dosage Form
- Product Age and Storage Conditions
1. Disintegration Time
- Disintegration is the process of mechanical breakdown of compressed tablets into small fragments after oral administration and it is the function of both amount of binder used and the compression force applied.
- It is of particular importance in case of solid dosage forms like tablets and capsules.
- It is an important aspect to ensure and maximize bioavailability.
- But, it does not guarantee the bioavailability of drugs because the fragmented particles must have to dissolve in fluid and then the only absorption is possible.
- The disintegration process assists the dissolution process, faster the disintegration- faster will be the dissolution, and increase the absorption rate and hence maximum bioavailability is achieved.
- If the amount of binders is higher, the longer will be the disintegration time, and the higher the compression force, the longer will be the disintegration time.
- So, if rapid disintegration is needed for therapeutic success, the disintegrating agents are used to ensure rapid disintegration time.
- After disintegration of a solid dosage form into granules, the granules must deaggregate into fine particles, as dissolution from such tiny particles is faster than that from granules.
2. Manufacturing/Processing Variables
- Drug dissolution is the single most important factor in the absorption of drugs, especially from the most widely used conventional solid dosage forms,- tablets and capsules.
- The dosage form related factors that influence dissolution and hence absorption of a drug from such formulations are:
- Excipients, and
- Manufacturing processes.
- The influence of excipients such as binders, lubricants, disintegrants, etc. on drug dissolution will be discussed in the subsequent section of this chapter currently we shall discuss influence of manufacturing processes.
- The processing factor of importance in the manufacture of capsules that can influence its dissolution is the intensity of packing of capsule contents.
- Several manufacturing processes influence drug dissolution from tablets are:
- Method of granulation, and
- Compression force.
Method of Granulation:
- There are two convenient types of granulation methods,
a. Wet granulation method and b. Dry granulation method.
- The wet granulation process is the mostconventional technique in the manufacture of tablets compared to other granulation methods, But, the limitations of this method include—
- The liquid may act as a medium for affecting chemical reactions such as hydrolysis, and
- The drying step may harm the thermolabile drugs.
- Involvement of large number of steps each of which can influence drug dissolution— i.e., moisture content, time and temperature of drying, duration of blending, etc.
- Dry granulation– The method of direct compression has been utilized to yield tablets that dissolve at a faster rate. But the hardness of the direct compressed tablet is less compared to the wet granulated and compressed tablets.
- Drugs which are moisture sensitive and the drugs which are thermolabile are formulated with dry granulation or direct compression method.
- Compression Force: The compression force employed in tabletingprocess influence density, porosity, hardness, disintegration time and dissolution of tablets. The curve obtained by plotting compression force versus rate of dissolution can take one of the 4 possible shapes shown in below Fig.
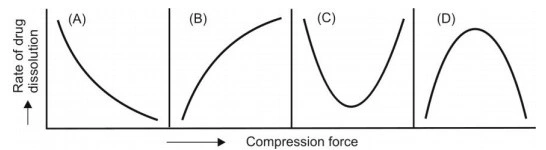
Fig. Influence of compression force on dissolution rate of tablets
- (A) On the one hand, higher compression force increases the density and hardness of tablet, decreases porosity and hence decreases the penetrability of the solvent into the tablet, retards wettability by forming a firmer and more effective sealing layer by the lubricant. In many cases, it promotes tighter bonding between the particles, all of which result in slowing of the dissolution rate of tablets (curve A of Fig. 2.25).
- (B) On the other hand, higher compression forces cause deformation, crushing or fracture of drug particles into smaller ones or convert a spherical granule into a disc shaped particle with a large increase in the effective surface area. This results in an increase in the dissolution rate of the tablet (curve B of Fig. 2.25).
- A combination of both the curves A and B is also possible as shown in curves C and D. In short, the influence of compression force on the dissolution rate is difficult to predict and a thorough study on each formulation should be made to ensure better dissolution and bioavailability.
- Intensity of Packing of Capsule Contents: Like the compression forcefor tablets, packing density in case of capsule dosage form can either inhibit or promote dissolution. Diffusion of GI fluids into the tightly filled capsules creates high pressure within the capsule resulting in rapid bursting and dissolution of contents. Opposite is also possible. It has been shown that capsules with finer particles and intense packing have poor drug release and dissolution rate due to a decrease in pore size of the compact and poor penetrability by the GI fluids.
3. Pharmaceutical Ingredients/Excipients (Formulation factors)
- A drug is rarely administered in its original form. Almost always, a convenient dosage form to be administered by a suitable route is prepared. Such a formulation contains a number of excipients.
- Excipients are added to ensure acceptability, physicochemical stability during the shelf-life, uniformity of composition and dosage, and optimum bioavailability and functionality of the drug product.
- Despite their inertness and utility in the dosage form, excipients can influence absorption of drugs.
- The more the number of excipients in a dosage form, the more complex it is and greater the potential for absorption and bioavailability problems.
- Commonly used excipients in various dosage forms are vehicles, diluents (fillers), binders and granulating agents, disintegrants, lubricants, coatings, suspending agents, emulsifiers, surfactants, buffers, complexing agents, colorants, sweeteners, crystal growth inhibitors, etc.
- Diluents (Fillers):
- Diluents are commonly added to tablet (and capsule) formulations if the required dose is inadequate to produce the necessary bulk.
- A diluent may be organic or inorganic. Among organic diluents, carbohydrates are very widely used—for example, starch, lactose, microcrystalline cellulose, etc. These hydrophilic powders are very useful in promoting the dissolution of poorly water-soluble, hydrophobic drugs like spironolactone by forming a coat onto the hydrophobic surface of drug particles and rendering them hydrophilic.
- Among the inorganic diluents, dicalcium phosphate (DCP) is most common. One classic example of drug-diluent interaction resulting in poor bioavailability is that of tetracycline and dicalcium phosphate. The cause is formation of divalent calcium-tetracycline complex which is poorly soluble and thus, unabsorbable.
- Binders and Granulating Agents:
- These materials are used to hold powders together to form granules or promote cohesive compacts for directly compressible materials and to ensure that the tablet remains intact after compression. Popular binders include polymeric materials (natural, semisynthetic and synthetic) like starch, cellulose derivatives, acacia, PVP, etc. Others include gelatin and sugar solution.
- In general, like fillers, the hydrophilic (aqueous) binders show better dissolution profile with poorly wettable drugs like phenacetin by imparting hydrophilic properties to the granule surface. However, the proportion of strong binders in the tablet formulation is very critical. Large amounts of such binders increase hardness and decrease disintegration/dissolution rates of tablets.
- PEG 6000 was found to be a strong binder for phenobarbital as it forms a poorly soluble complex with the drug.
- Non-aqueous binders like ethyl cellulose also retard drug dissolution.
- Disintegrants:
- These agents overcome the cohesive strength of tablets and break them up on contact with water which is an important prerequisite to tablet dissolution. Almost all the disintegrants are hydrophilic in nature. A decrease in the amount of disintegrant can significantly lower bioavailability.
- Adsorbing disintegrants like bentonite and veegum should be avoided with low dose drugs like digoxin, alkaloids and steroids since a large amount of dose is permanently adsorbed and only a fraction is available for absorption.
- Microcrystalline cellulose is a very good disintegrant (and a binder too) but at high compression forces, it may retard drug dissolution.
- Lubricants:
- These agents are added to tablet formulations to aid flow of granules, to reduce interparticle friction and sticking or adhesion of particles to dies and punches. The commonly used lubricants are hydrophobic in nature and known to inhibit wettability, penetration of water into tablet and their disintegration and dissolution. This is because the disintegrant gets coated with the lubricant if blended simultaneously which however can be prevented by adding the lubricant in the final stage.
- The best alternative is use of soluble lubricants like SLS and carbowaxes which promote drug dissolution.
- Coatings:
- In general, the negative effect of various coatings on drug dissolution from a tablet dosage form is in the following order:
- Enteric coat > Sugar coat > Non-enteric film coat.
- The dissolution profile of certain coating materials change on aging; e.g. shellac coated tablets, on prolonged storage, dissolve more slowly in the intestine. This can, however, be prevented by incorporating little PVP in the coating formulation.
4. Nature and Type of Dosage Form
- Apart from the proper selection of drug, clinical success often depends to a great extent on the proper selection of dosage form of that drug. For a given drug, a 2 to 5 fold or perhaps more difference could be observed in the oral bioavailability of a drug depending upon the nature and type of dosage form.
- Such a difference is due to the relative rate at which a particular dosage form releases the drug to the biological fluids and the membrane.
- The more complex a dosage form, greater the number of rate-limiting steps and greater the potential for bioavailability problems.
- The rate at which a particular dosage form releases the drug following administration is given in Below Fig.
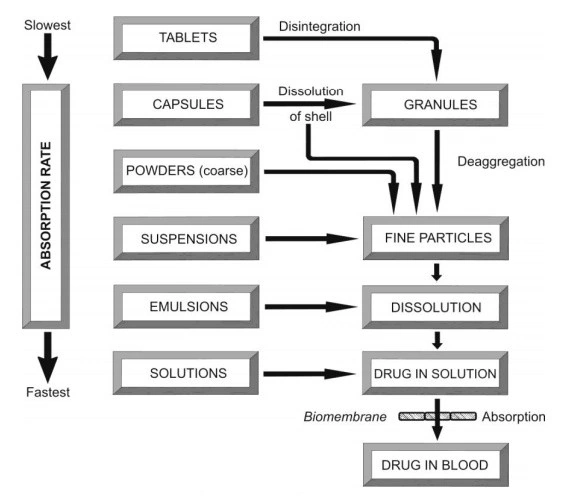
Fig. Course of events that occur following oral administration of various dosage forms
- As a general rule, the bioavailability of a drug from various dosage forms decreases in the following order:
Solutions > Emulsions > Suspensions > Capsules > Tablets > Coated Tablets > Enteric Coated Tablets > Sustained Release Products.
- Thus, absorption of a drug from solution is fastest with least potential for bioavailability problems whereas absorption from a sustained release product is slowest with greatest bioavailability risk.
- Solutions: A drug in a solution (syrups, elixirs, etc.) is most rapidlyabsorbed since the major rate-limiting step, drug dissolution, is absent. Factors that influence bioavailability of a drug from solution dosage form include—the nature of solvent (aqueous, water miscible, etc.), viscosity, surfactants, solubilisers, stabilizers, etc. Quite often, dilution of a drug in solution with GI fluids results in precipitation of drug as fine particles which generally dissolve rapidly. Factors that limit the formulation of a drug in solution form include stability, solubility, taste, cost of the product, etc.
- Emulsions: Emulsion dosage forms have been found to be superior tosuspensions in administering poorly aqueous soluble lipophilic drugs. It was observed with indoxole (an NSAID) that when it is dissolved in a vegetable oil and emulsified in water, absorption increases 3 fold over its aqueous suspension. Emulsion dosage form presents a large surface area of oil to the GIT for absorption of a drug.
- Suspensions: The major rate-limiting step in the absorption of a drug fromsuspension dosage form is drug dissolution which is generally rapid due to the large surface area of the particles. Important factors in the bioavailability of a drug from suspensions include particle size, polymorphism, wetting agents, viscosity of the medium, suspending agents, etc.
- Powders: Though powders are superior to tablets and capsules, they arenot in use nowadays due to handling and palatability problems. Major factors to be considered in the absorption of a drug from powders are particle size, polymorphism, wettability, etc.
- Capsules: Powders and granules are popularly administered in hard gelatincapsules whereas viscous fluids and oils in soft elastic shells. Factors of importance in case of hard gelatin capsules include drug particle size, density, polymorphism, intensity of packing and influence of diluents and excipients. Hydrophilic diluents like lactose improve wettability, deaggregation and dispersion of poorly aqueous soluble drugs whereas inhibitory effect is observed with hydrophobic lubricants like magnesium stearate. Soft elastic capsules as such dissolve faster than hard gelatin capsules and tablets and show better drug availability from oily solutions, emulsions or suspensions of medicaments.
- Tablets: Compressed tablets are the most widely used convenience and cost effective dosage forms. A schematic representation of disintegration, deaggregation, dissolution and absorption of a drug from a tablet dosage form is shown in Below Fig.
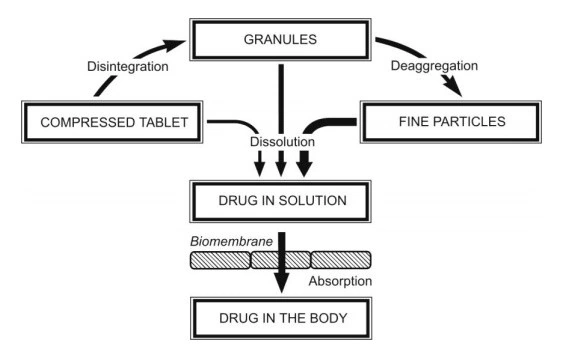
Fig. Sequence of events in the absorption of a drug from tablet dosage form
- In tablets, As a general rule, the bioavailability of a drug from various dosage forms decreases in the following order:
Effervescent Tablets > Sublingual/Buccal Tablets > Immediate-Release Tablets> Film-Coated Tablets > Enteric-Coated Tablets > Extended/Modified-Release Tablets
5. Product Age and Storage Conditions
- A number of changes, especially in the physicochemical properties of a drug in dosage form, can result due to aging and alterations in storage conditions which can adversely affect bioavailability.
- With solution, suspension and emulsion dosage form, precipitation, sedimentation and cake formation may occur during storage.
- Changes that occur during the shelf-life of a dosage form are affected mainly by large variations in temperature and humidity. In one of the studies conducted on prednisone tablets containing lactose as the filler, high temperature and high humidity resulted in harder tablets that disintegrated and dissolved slowly.
- Temperature: According to Arrhenius theory, increase in temperature leads to an increase in the rate of reaction, thus leads to degradation and formation of harmful metabolites resulting in a decrease in bioavailability.
- pH: Change in pH results in a change in the rate of decomposition of drugs. Most of the drugs are stable at a particular pH only (4-8 pH).
- Moisture: Water acts as a catalyst for many chemical reactions such as oxidation and hydrolysis. It promotes the growth of microorganisms.
- Incompatibilities: An interaction between the excipient and drug in a dosage form may sometime lead to the instability of the product.
- Many other factors like light, oxygen, type of packaging and type of dosage form may also affect drug stability.
BIOLOGICAL or PATIENT RELATED FACTORS AFFECTING DRUG ABSORPTION
- Age
- Gastric emptying time
- Intestinal transit time
- Gastrointestinal pH
- Disease states
- Blood flow through the GIT
- Gastrointestinal contents:
- Other drugs
- FoodFluids
- Fluids
- Other normal GI contents
- First pass metabolism/ Presystemic metabolism by:
- Luminal enzymes.
- Gut wall enzymes
- Bacterial enzymes
- Hepatic enzymes
Before dealing with the patient related factors influencing bioavailability of a drug, the anatomy and physiology of the gastrointestinal tract will be discussed briefly.
Gastrointestinal tract
- The gastrointestinal tract (GIT) comprises of a number of components, their primary function being secretion, digestion and absorption.
- The total length of the gastrointestinal tract (GIT) in a normal, living adult is approximately 5 to 9 meters (roughly 16 to 30 feet) when measured from the mouth to the anus.
- The major functional components of the GIT are stomach, small intestine (duodenum, jejunum and ileum) and large intestine (colon) which grossly differ from each other in terms of anatomy, function, secretions and pH. (Below Fig. and Table).
- The entire length of the GI mucosa from stomach to large intestine is lined by a thin layer of mucopolysaccharides (mucus/mucin).
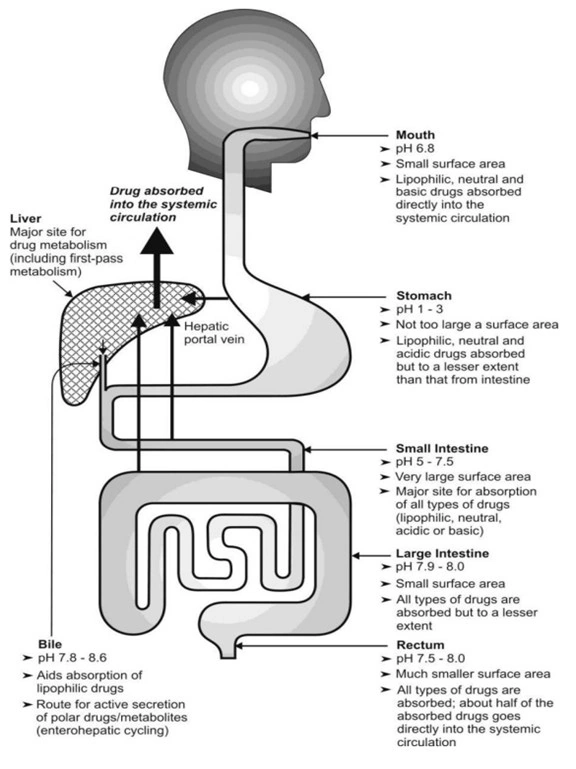
Fig. Schematic representation of the GIT and different sites of drugabsorption
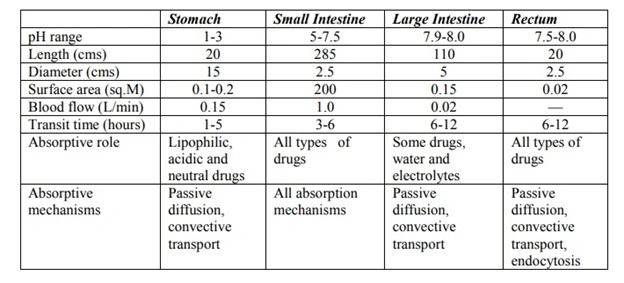
TABLE: Anatomical and Functional Differences Between the Important Regions of the GIT
- Stomach: The stomach is a bag like structure having a smooth mucosa andthus small surface area. Its acidic pH, due to secretion of HCl, favours absorption of acidic drugs, since they are unionised to a large extent in such a pH. But not aid the dissolution of the acidic drug. The gastric pH aids dissolution of basic drugs due to salt formation and subsequent ionisation which are therefore absorbed to a lesser extent from stomach because of the same reason. The stomach is not the principal region for drug absorption because – The total mucosal area is small and The epithelium is dominated by mucus-secreting cells rather than absorptive cells.
- Small Intestine: It is the major site for absorption of most drugs due to itsspecial characteristics –
- Large surface area –the folds in the intestinal mucosa, called as the folds of Kerckring, result in 3 fold increase in the surface area. The surface of these folds possess finger like projections called as villi which increase the surface area 30 times. From the surface of villi protrude several microvilli (about 600 from each absorptive cell that lines the villi) resulting in 600 times increase in the surface area (Fig.).
- Great length of small intestine (approximately 285 cm.)–result in more than 200 square meters of surface which is several times that of stomach.
- Greater blood flow – the blood flow to the small intestine is 6 to 10times that of stomach.
- Favourable pH range –the pH range of small intestine is 5 to 7.5which is favourable for most drugs to remain unionised.
- Slow peristaltic movement –prolongs the residence time of drug in the intestine.
- High permeability – the intestinal epithelium is dominated by absorptive cells.
A contribution of all the above factors thus make intestine the best site for absorption of most drugs.
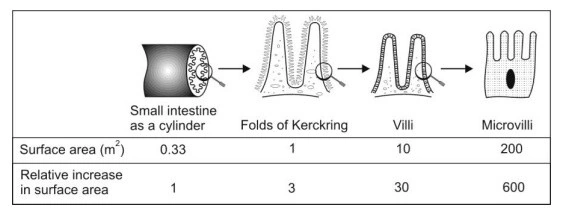
Fig. Representation of the components of intestinal epithelium that accounts for its large surface area
- Large Intestine: Its length and mucosal surface area is very small (villi and microvilli are absent) in comparison to small intestine and thus absorption of drugs from this region is insignificant. Its contents are neutral or alkaline. The main role of large intestine is in the absorption of water and electrolytes. However, because of the long residence time (6 to 12 hours), colonic transit may be important in the absorption of some poorly soluble drugs and sustained release dosage forms.
BIOLOGICAL or PATIENT RELATED FACTORS AFFECTING DRUG ABSORPTION:
1. Age
- The oral dosage form (tablets/capsules) is designed for the full release of the drug over a specific period of time.
- In infants, the gastric pH is high, intestinal surface is low and blood flow to the GIT is low resulting in altered absorption pattern in comparison to adults.
- In elderly people, causes of impaired drug absorption because of loss of several microvilli on the mucosal intestinal surface, reduction in GI blood flow and decreases gastric acidity, higher incidents of achlorhydria and bacterial overgrowth in small intestine.
2. Gastric emptying
- The passage of gastric content from the stomach to the small intestine is known as gastric emptying, and the major site of drug absorption is intestine. Thus, generally speaking, rapid gastric emptying increases bioavailability of a drug.
- At an empty stomach, drug absorption is higher than a full stomach.
- If the gastric emptying time prolongs, drug may degrade in gastric pH and affects its bioavailability.
- Rapid gastric emptying is advisable where:
- A rapid onset of action is desired e.g. sedatives.
- Dissolution of drug occurs in the intestine e.g. enteric-coated dosage forms.
- The drugs are not stable in the gastric fluids e.g. penicillin G and erythromycin.
- The drug is best absorbed from the distal part of the small intestine e.g. vitamin B12.
- For better dissolution and absorption, the gastric emptying can be promoted by taking the drug on empty stomach.
- Delay in gastric emptying is recommended in particular where:
- The food promotes drug dissolution and absorption e.g. griseofulvin.
- Disintegration and dissolution of dosage form is promoted by gastric fluids.
- The drugs are absorbed from the proximal part of the small intestine and prolonged drug-absorption site contact is desired e.g. vitamin B2 and vitamin C
- Gastric emptying time may alter due to several factors such as :
- Volume of a meal: Larger the bulk of the meals, longer the gastric emptying time.
- Composition of meal: Predictably, the rate of gastric emptying for various food materials is in the following order: carbohydrates > proteins > fats.
- Physical state and viscosity of meal: Liquid meals take less than an hour to empty whereas a solid meal may take as long as 6 to 7 hours. Viscous materials empty at a slow rate in comparison to less viscous materials.
- GI pH: Gastric emptying is retarded at low or acidic stomach pH and promoted at higher or alkaline pH.
- Body posture: Gastric emptying is favoured while standing and by lying on the right side since the normal curvature of the stomach provides a downhill path whereas lying on the left side or in supine position retards it.
- Emotional state: Stress and anxiety promote gastric motility whereas depression retards it.
- Disease state: Diseases like gastroenteritis, gastric ulcer, pyloric stenosis, diabetes and hypothyroidism retard gastric emptying. Partial or total gastrectomy, duodenal ulcer and hyperthyroidism promote gastric emptying rate.
- Drugs: Drugs that retard gastric emptying include poorly soluble antacids (aluminium hydroxide), anticholinergics (atropine, propantheline), narcotic analgesics (morphine) and tricyclic antidepressants (imipramine, amitriptyline). Domperidone like drugs stimulate gastric emptying.
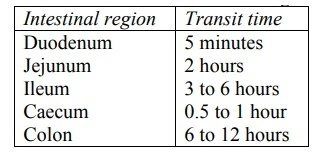
3. Intestinal transit time
- Intestinal transit (or gastrointestinal transit) is the movement of food, liquids, and waste through the digestive tract—from the stomach through the small and large intestines (colon) to the rectum and the time taken is known as intestinal transit time. The small intestine provides a large surface area for the absorption of drugs, the longer residence time of drug therefore achieve complete drug absorption.
- Here peristaltic movement plays an important role in drug absorption. The residence time depends upon the intestinal motility or contractions.
- Delayed intestinal transit is desirable for:
- Drugs that dissolve or release slowly from their dosage form (sustained-release products).
- Drugs that dissolve only in the intestine (enteric-coated formulations).
- Drugs which are absorbed from specific sites in the intestine (several B vitamins, lithium carbonate, etc.).
- When the drug penetrates the intestinal mucosa very slowly e.g. acyclovir.
- When absorption of drug from the colon is minimal.
4. Gastrointestinal pH
- As studied earlier, most of the drugs are weak acid and weak bases. The lipophilic membrane is impermeable to an ionized drug.
- It is well known that the weakly acidic drugs are absorbed in acidic pH (acidic pH 1.4 – 2) i.e., in the stomach at a faster rate and weakly basic drugs are well absorbed in basic pH (7.5 – 8) i.e. in the intestine at a faster rate.
- The acidic drugs are not ionized in an acidic medium and the basic drugs are not ionized in a basic medium so that the Unionized drugs can pass the membrane easily.
5. Disease states
- The Disease States may lower or higher the absorption rate as disease state effects Gastric emptying and Intestinal transit time.
- The anatomy of the GI track may change due to GI surgery and alter the absorption process.
- Some of the disease and impacts on drug absorption:
| Disease State | Primary Impact on Absorption |
| Constipation | Increase the drug absorption as it increases the intestinal transit time. |
| Diarrhoea | Decrease the drug absorption as it decreases the intestinal transit time. |
| Celiac Disease | Decreased surface area; decreases the rate of drug absorption. |
| Heart Failure | Reduced intestinal blood flow and decrease in drug bioavailability. |
| Liver Cirrhosis | Increased bioavailability of oral drugs due to reduced first-pass metabolism. |
6. Blood flow through GIT
- GIT is composed of a blood capillary network and blood flow to this region is very high then the other part of the body.
- Because of highly perfumes tissue is lined on the surface of whole GIT that ensures high drug absorption to these regions.
- The high perfusion rate of GIT ensures that once the drug has crossed the membrane, it is rapidly removed from the absorption site thus maintaining the sink conditions and concentration gradient for continued drug absorption.
- Higher the blood flow to GIT, more the absorption of drug and Lower the blood flow to GIT, lesser the absorption of drug.
7. Gastrointestinal Contents
A number of GI contents can influence drug absorption as discussed below:
- Food-drug interactions: Presence of food may either delay, reduce, increase or may not affect drug absorption. (Table 2.9).
| Food/Drink | Affected Drug(s) | Impact on Absorption |
| High-fat Meal | Lipophilic drugs (e.g., Saquinavir) | Increases absorption due to bile salt solubilization. |
| Dairy Products | Tetracycline, Ciprofloxacin | Decreases absorption via calcium binding. |
| Grapefruit Juice | Statins, Felodipine | Increases absorption by inhibiting gut metabolism. |
| Orange Juice | Fexofenadine, Celiprolol | Decreases absorption by inhibiting uptake transporters. |
- Fluid volume: Administration of a drug with large fluid volume results in better dissolution, rapid gastric emptying and enhanced absorption—for example, erythromycin is better absorbed when taken with a glass of water under fasting condition than when taken with meals.
- Interaction of drug with normal GI constituents: The GIT containsa number of normal constituents such as mucin, bile salts and enzymes which influence drug absorption.
- Mucin, a protective mucopolysaccharide that lines the GI mucosa, interacts with some drugs and retards their absorption as it acts as a barrier to diffusion of drugs.
- The bile salts aid solubilisation and absorption of lipid soluble drugs like griseofulvin and vitamins A, D, E and K on one hand decreases the solubilisation of hydrophilic drugs.
- Drug-Drug interactions in the GIT: Like food-drug interactions, drug-drug interactions can either be physicochemical or physiological.
| Interaction Type | Example “Interfering” Drug | Effect on Second Drug |
| pH Change | Omeprazole (PPI) | Decreased absorption of acidic drugs. |
| Chelation | Antacids (Al/Mg) | Decreased absorption of Ciprofloxacin. |
| Motility | Morphine (Opioid) | Delayed onset of action for most oral meds. |
| Transporter | Quinidine | Increased absorption of Digoxin. |
| Competition for Transporters | Verapamil | Competes for carrier mediated diffusion |
8. Presystemic Metabolism/First-Pass Effects
- Drug metabolized in the body results in a decrease in the concentration of active drug this phenomenon is called as the first-pass metabolism or pre systemic metabolism.
- First-pass/presystemic metabolism is one of the reasons for decreased bioavailability of oral dosage forms. (see figure 2.30)
- For example, after oral administration of propranolol, it shows 26 % bioavailability, as 75-85% drug metabolism before it reaches the systemic circulation.
- Hence, to aid the first-pass effect and to achieve the desired concentration in the systemic circulation, the dose of the drug has to increase.
- But in the case of liver disease, metabolism of some drug is decrease and increase the bioavailability that leads to toxic effect. Thus it is necessary to decrease the dose amount in liver diseases.
- The enzymes that affect the presystemic metabolism and decrease the bioavailability of the drug are:
- Luminal enzymes (intestinal and pancreatic)
- Gut wall enzymes (mucosal enzymes)
- Bacterial enzymes (colonic enzymes)
- Hepatic enzymes (liver)
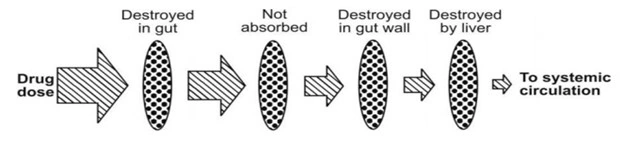
Fig. Processes that reduce the availability of orally administered drugs
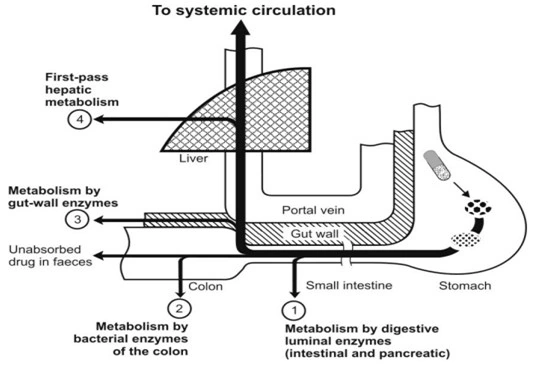
Fig. Different sites of presystemic metabolism
Absorption of drug from Non per oral extra-vascular routes
- Except IV route, all other routes of administration involve an absorption step including oral, intramuscular, subcutaneous, nasal, ophthalmic, rectal, vaginal, sublingual and pulmonary.
- These routes not only bypasses the GIT absorption and reached systemically but also these routes bypasses the hepatic first-pass effect and enzymatic degradation.
- Therapeutic agents like proteins and peptides can be administered by the extravascular route.
- Various drug delivery systems have been researched to offer maximum absorption and bioavailability such as,
- Buccal sublingual drug delivery
- Rectal drug delivery
- Transdermal drug delivery
- Intramuscular injection
- Subcutaneous injection
- Pulmonary drug delivery
- Nasal drug delivery
- Ocular drug delivery
- Vaginal drug delivery
- Various non per oral extravascular routes are discussed as follows:
1. Buccal Sublingual Route
- Buccal mucosa is a potential site for the absorption of drugs administered by the buccal route for systemic and local effects.
- This route is particularly interesting because it bypasses the hepatic first-pass effect and enzymatic degradation. The buccal root provides a large area for absorption as compared to the nose, vagina and rectum.
- Oral mucosa is more permeable to proteins and peptide molecules and has a higher perfusion rate than the skin, hence the absorption rate is higher for the larger size of drug
- Factors affecting buccal and sublingual drug delivery.
- Lipophilicity of drug- require high lipid soluble for passive diffusion.
- Salivary secretion-required for drug dissolution and diffusion.
- pH of saliva-adult clean pH drug remains unionized
- Oral mucosal epithelium– rate-limiting factor.
- Categories of drugs administered by this route are antianginal, antihypertensive, analgesics, antacids and many proteins and peptides.
- Dosage forms available – tablets, films, buccal patch, gel, paste, ointment.
2. Rectal Route
- This route is a choice for a patient like unconscious and vomiting children. The local and systemic effects can be achieved by the rectal route.
- This route is useful for the drug which shows limited or poor absorption at the upper part of the GI tract, a drug which is degraded in proteolytic enzymes, drugs which show a high rate of hepatic first-pass effect and the drug which are irritated to the gastric mucosa.
- Factors that restrict the use of the rectal route are irritation and the presence of fecal matter.
- Dosage forms available are suppositories, pessaries and gels.
3. Transdermal Route
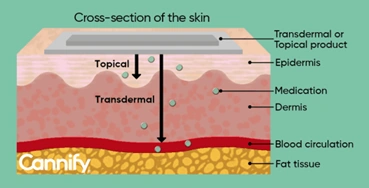
- The objective of the transdermal route of administration is to deliver the drug into systemic circulation through the skin membrane by avoiding the first-pass effect. The transdermal route is a good substitute for the oral route due to minimal side effects and unconditional intestinal incompatibility.
- Termination of therapy is possible at any time.
- Anatomically the skin has three layers like the epidermis, dermis, and subcutaneous tissue.
- The outer layer epidermis is a rate-limiting step in diffusion of the drug because it is composed of nonvascular, multilayered cellular networks and the dermis allows the drug to penetrate through the skin because of its composed of highly vascular tissues and the drug is taken up into systemic circulation.
- Factors affecting percutaneous absorption are the thickness of the epidermis, hair follicles, skin hydration, skin condition, and permeation enhancers. Examples of drugs given by the transdermal route are scopolamine (motion sickness), nitroglycerine (angina pectoris), clonidine (hypertension) estradiol (menopausal symptoms).
5. Subcutaneous Injection
- Under the skin injection is one of the alternative methods used to deliver medication. A short needle is used to inject the medication between muscle and skin. Subcutaneous injections absorb slowly than the intravenous injection over a period of 24 hours. Those medications which are unstable at acidic pH and enzymatic degradation in GIT are given by this route.
- This injection has particular importance when a rapid response is not desired and to avoid the first-pass effect. Factors that restrict the use of subcutaneous injection are the volume of injection should be less than 1 ml but in some cases 2 ml is safe. Examples of drug administered through subcutaneous injection are epinephrine, morphine, hydro morphine, metoclopramide, and dexamethasone.
6. Pulmonary Route
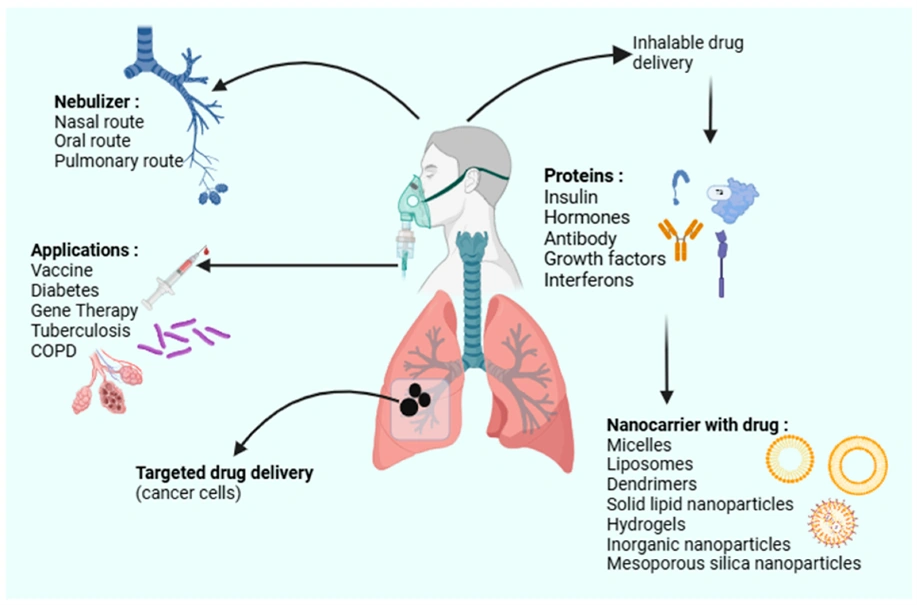
- The lung is the only organ that passes the entire correct output of a heart ventricle.
- A thin alveolar vascular permeable barrier is responsible for drug absorption.
- Certain anatomical properties like 600 million alveoli, enormous epithelial surface area and single cellular epithelium efficiently assist drug transport.
- Lipophilic drugs pass through passive transport and hydrophilic drug by pore transport.
- Pulmonary root has certain advantages like
- Site-specific drug delivery can be achieved.
- Systemic side effects are minimized.
- Rapid onset of action.
- Avoid GI absorption, hence bypasses the first-pass effect.
- Therapeutic equivalence to that of drug given by oral route.
- Factors that affect pulmonary effectiveness are mucociliary clearance, moisture, airways geometry, and alveolar macrophages.
- Examples of drug used via pulmonary route: Salbutamol, beclomethasone, chromaline, and certain proteins and peptides.
7. Internasal Route
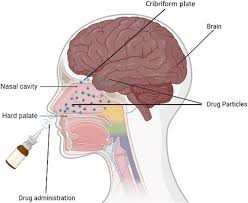
- The nasal route is one of the alternative routes which provides vascularized mucosa for systemic and local delivery of a drug.
- Absorption of drugs in the nasal cavity follows the diffusion pathway via the mucosal surface followed by penetration, permeation, and absorption.
- The olfactory region provides direct Access to CNS, so drugs can be delivered by the intracellular and extracellular pathway to the CNS.
- A most popular drug like anti-inflammatory and topical decongestants can be delivered by the nasal route.
- Some drugs have direct access to systemic absorption by avoiding the first-pass effect.
- Factors affecting drug transport through the nasal route: Molecular weight, the concentration of drug, dose, the volume of administration and mucociliary clearance.
8. Intraocular Route
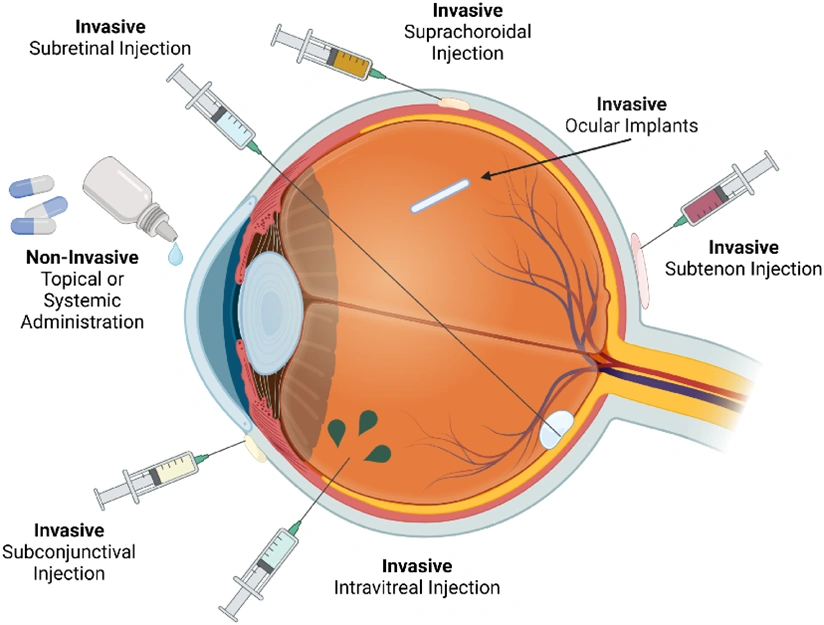
- It is one of the alternative routes to deliver the drug locally and systematically. Topical administration is employed to treat the anterior part of the eye example cornea, conjunctiva, sclera, iris, and ciliary body.
- Major factors which limit the residence time of drug are loss on drainage due to blinking, tear turnover, tear film, and lacrimation. Cornea act as a barrier to drug penetration and absorption. Systemic administration is employed to treat the posterior segment of the eye.
- For satisfactory absorption of the drug, the drug has to pass the blood aquarius barrier and blood-retinal barrier, which acts as a rate-limiting step.
- Drug-like mydriatic, meiotic, and local anesthetics are delivered through the eye or used to treat eye syndromes and diseases.
9. Vaginal Route
- Absorption of a drug from the vagina generally occurs in two-step
- 1) Dissolution of the drug in vaginal lumens 2) Membrane permeation by passive diffusion.
- This is one of the alternative route for the drug delivery whose activity in GIT decreases due to enzymatic degradation for the first pass effect. The absorption rate of drugs after vaginal administration may vary due to patient age, menstrual cycle, vaginal physiology and formulation factors.
- The original surface consists of an epithelial layer, a muscular coat, and the tunica adventia.
- The thickness of the vaginal epithelium changes during the menstrual cycle, which limits the effective drug absorption, also the vaginal secretion (amount and composition) may vary during the menstrual cycle can also affect the bioavailability.
- Commonly used dosage forms are tablets, gel, cream, suppositories and vaginal rings, etc.
Distribution of Drugs
- After entry into the systemic circulation, either by intravascular injection or by absorption from any of the various extravascular sites, the drug is subjected to a number of processes called as disposition processes.
- Disposition is defined as the processes that tend to lower the plasma concentration of drug. The two major drug disposition processes are–
- Distribution which involves reversible transfer of a drug between compartments.
- Elimination which causes irreversible loss of drug from the body. Elimination is further divided into two processes –
- Biotransformation (metabolism)
- Excretion.
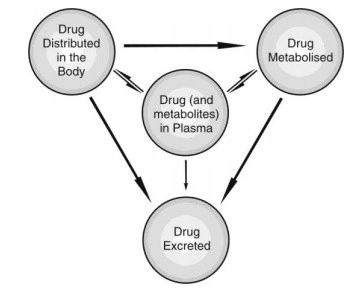
Fig. 32. Interrelationship between different processes of drug disposition.
- As stated above, distribution is defined as the reversible transfer of a drug between one compartment and another. Since the process is carried out by the circulation of blood, one of the compartments is always the blood or the plasma and the other represents extravascular fluids and other body tissues. In other words, distribution is reversible transfer of a drug between the blood and the extravascular fluids and tissues.
- Distribution is a passive process,for which, the driving force is concentration gradient between the blood and the extravascular tissues. The process occurs by diffusion of free drug only until equilibrium is achieved.
- As the pharmacological action of a drug depends upon its concentration at the site of action, distribution plays a significant role in the onset, intensity and sometimes duration of drug action.
Steps in Drug Distribution
- Before studying steps in drug distribution, we shall see what is extracellular/ interstitial fluid (ECF) and Intracellular fluid (ICF). (Fig. 33).
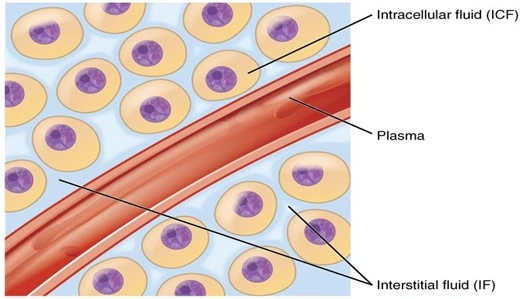
Fig. 33. extracellular/ interstitial fluid (ECF) and Intracellular fluid (ICF).
Steps
- Distribution of drug present in systemic circulation to extravascular tissues involves following steps (Fig.34.)-
- Permeation of free or unbound drug present in the blood through the capillary wall (occurs rapidly) and entry into the interstitial/extracellular fluid (ECF).
- Permeation of drug present in the ECF through the membrane of tissue cells and into the intracellular fluid. This step is rate-limiting and depends upon two major factors –
- Rate of perfusion to the extracellular tissue
- Membrane permeability of the drug.
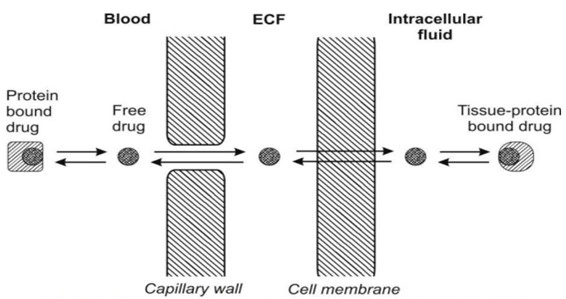
Fig. 34. Schematic of the step involved in drug distribution.
Factors Affecting Distribution of Drugs
- Distribution of a drug is not uniform throughout the body because different tissues receive the drug from plasma at different rates and to different extents. Differences in drug distribution among the tissues essentially arise as result of a number of factors as enumerated below –
- Tissue permeability of the drug:
- Physicochemical properties of the drug like molecular size, pKa and o/w partition coefficient
- Physiological barriers to diffusion of drugs (Simple capillary endothelial barrier, Simple cell membrane barrier, Blood-brain barrier, Blood-CSF barrier, Blood- placental barrier, and Blood-testis barrier.)
- Organ/tissue size and perfusion rate.
- Binding of drugs to tissue components:
- Binding of drugs to blood components
- Binding of drugs to extravascular tissue proteins
- Miscellaneous factors:
- Age
- Pregnancy
- Obesity
- Diet
- Disease states
- Tissue permeability of the drug:
1. Tissue permeability of the drug:
- Tissue permeability of a drug refers to its ability to pass through biological membranes and tissues.
- Tissue permeability of a drug depends upon the physicochemical properties of the drug as well as the physiological barriers that restrict diffusion of drug into tissues.
- Physicochemical properties of the drug like molecular size, pKa and o/w partition coefficient:
- Molecular size: Almost all drugs having molecular weight less than 600 Daltons easily cross the capillary membrane to diffuse into the interstitial/ extracellular fluids. Only small, water-soluble molecules of size below 50 Daltons enter the cell through aqueous filled channels. Whereas those of larger sizes are restricted unless a specialized transport system exists for them.
- pKa or degree of ionization: The degree of ionisation of a drug is an important determinant in its tissue penetrability. The pH of the blood and the extravascular fluid also play a role in the ionisation and diffusion of drugs into cells.
A drug that remains unionised at these pH values can permeate the cells relatively more rapidly. Since the blood and the ECF pH normally remain constant at 7.4, they do not have much of an influence on drug diffusion unless altered in conditions such as systemic acidosis or alkalosis.
All drugs that ionise at plasma pH (i.e. polar, hydrophilic drugs), cannot penetrate the lipoidal cell membrane and tissue permeability isthe rate-limiting step in the distribution of such drugs. - O/W partition coefficient: Only unionised drugs which are generally lipophilic, rapidly cross the cell membrane. Among the drugs that have same o/w partition coefficient but differ in the extent of ionisation at blood pH, the one that ionises to a lesser extent will have greater penetrability than that which ionises to a larger extent; for example, pentobarbital and salicylic acid have almost the same Ko/w but the salicylic acid is more unionised at blood pH and therefore distributes rapidly.
In case of polar drugs where permeability is the rate-limiting step in the distribution, the driving force is the effective partition coefficient of drug. More the partition coefficient, more the drug distribution and vise versa.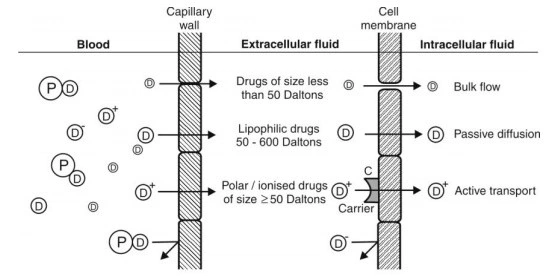
Fig. 35. Permeation of unionised and ionised drugs across the capillary and the cell membrane.
- Physiological barriers to diffusion of drugs:
A membrane (or a barrier) with special structural features can be a permeability restriction to distribution of drugs to some tissues. Some of the important simple and specialized physiological barriers are:- Simple capillary endothelial barrier.
- Simple cell membrane barrier.
- Blood-brain barrier.
- Blood-Cerebrospinal Fluid barrier.
- Blood- placental barrier.
- Blood-testis barrier.
- Physicochemical properties of the drug like molecular size, pKa and o/w partition coefficient:
- The Simple Capillary Endothelial Barrier:
The membrane of capillaries that supply blood to most tissues is, practically speaking, not a barrier to drugs. Thus, all drugs, ionised or unionised, with a molecular size less than 600 Daltons, diffuse through the capillary endothelium and into the interstitial/extracellular fluid. Only drugs bound to the blood components are restricted because of the large molecular size of the complex. - The Simple Cell Membrane Barrier:
Once a drug diffuses from the capillary wall into the extracellular fluid, its further entry into cells of most tissues is limited by its permeability through the membrane that lines such cells. Such a simple cell membrane is similar to the lipoidal barrier in the GI absorption of drugs. The physicochemical properties that influence permeation of drugs across such a barrier are summarized in Fig. 36.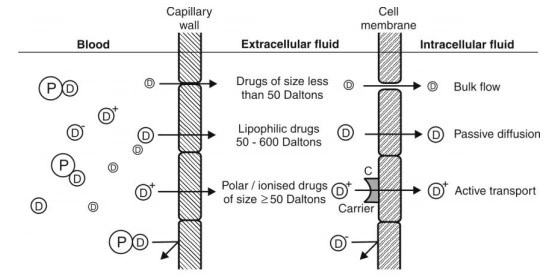
Fig. 36. Plasma membrane barrier and drug diffusion across it. - Blood-Brain Barrier (BBB):
- Unlike the capillaries found in other parts of the body, thecapillaries in the brain are highly specialized and much less permeable to water-soluble drugs.
- The brain capillaries consist of endothelial cells which are joined to one another by continuous tight intercellular junctions comprising what is called as the blood-brain barrier(Fig. 38.). Moreover, the presence of special cells called as pericytes and astrocytes, which are the elements of the supporting tissue found at the base of endothelial membrane, form a solid envelope around the brain capillaries. As a result, the intercellular (paracellular) passage is blocked and for a drug to gain access from the capillary circulation into the brain, it has to pass through the cells (transcellular) rather than between them.
- However, there are specific sites in the brain where the BBB does not exist, namely, the trigger area and the median hypothalamic eminence. hence, drugs administered intranasally may diffuse directly into the CNS. (Fig. 39.)
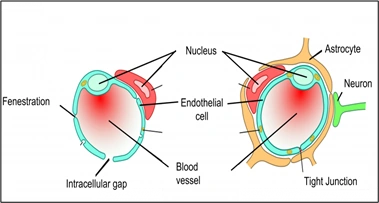
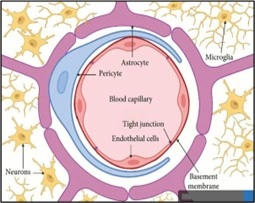
Fig. 38. pericytes and astrocytes
Fig. 39. Medan eminence
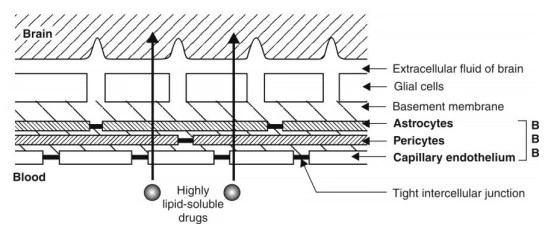
Fig. 40. Blood-brain barrier-CSF barrier
- A solute may thus gain access to brain via only one of two pathways:
- Passive diffusion through the lipoidal barrier – which is restricted to small molecules (molecular weight less than 700 Daltons) having high o/w partition coefficient.
- Active transport of essential nutrients such as sugars and amino acids. Thus, structurally similar foreign molecules can also penetrate the BBB by the same mechanism.
- The selective permeability of lipid soluble drug through the BBB makes appropriate choice of a drug to treat CNS disorders an essential part of therapy; for example, Parkinsonism, a disease characterized by depletion of dopamine in the brain, cannot be treated by administration of dopamine as it does not cross the BBB. Hence, levodopa, which can penetrate the CNS where it is metabolised to dopamine, is used in its treatment.
- Targeting of polar drugs to brain in certain conditions such as tumour had always been a problem. Three different approaches have been utilized successfully to promote crossing the BBB by drugs:
- Use of permeation enhancers such as dimethyl sulphoxide (DMSO).
- Osmotic disruption of the BBB by infusing internal carotid artery with mannitol.
- Use of dihydropyridine redox system as drug carriers to the brain.
- Blood-Cerebrospinal Fluid Barrier:
- The cerebrospinal fluid (CSF) is formed mainly by thechoroid plexus of the lateral, third and fourth ventricles (Fig. 41.) and is similar in composition to the ECF of brain.
- The capillary endothelium that lines the choroid plexus have open junctions or gaps and drugs can flow freely into the extracellular space between the capillary wall and the choroidal cells.
- However, the choroidal cells are joined to each other by tight junctions forming the blood-CSF barrier which has permeability characteristics similar to that of the BBB (Fig. 42.).
- Only highly lipid soluble drugs can cross the blood-CSF barrier with relative ease whereas moderately lipid soluble and partially ionised drugs permeate slowly.
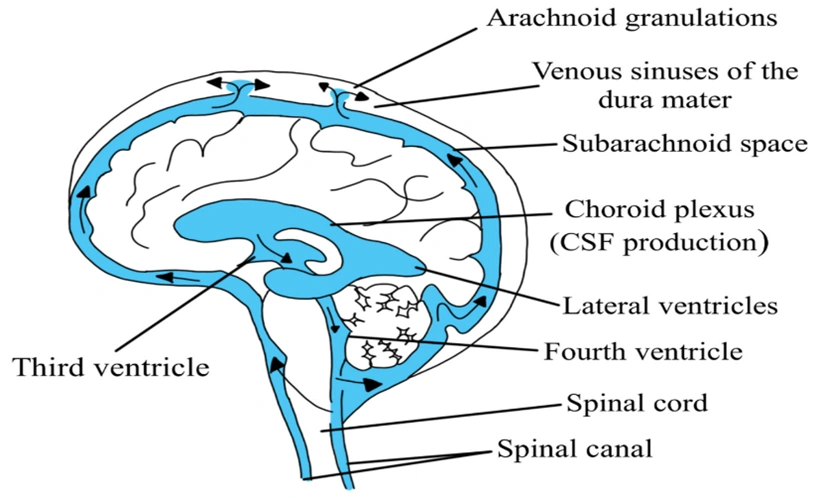
Fig. 41. The cerebrospinal fluid (CSF)
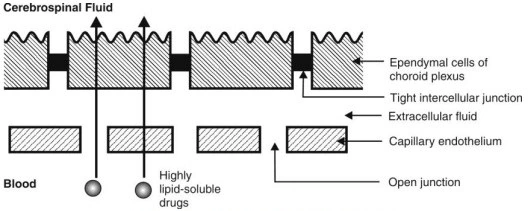
Fig. 42. The blood-CSF barrier
- Blood-Placental Barrier:
- The maternal and the foetal blood vessels are separated by anumber of tissue layers made of foetal trophoblast basement membrane and the endothelium which together constitute the placental barrier. The flow of blood in the maternal and the foetal blood vessels is shown in Fig. 43.
- The human placental barrier has a mean thickness of 25 microns in early pregnancy that reduces to 2 microns at full term which however does not reduce its effectiveness. Many drugs having molecular weight less than 1000 Daltons and moderate to high lipid solubility e.g. ethanol, sulphonamides, barbiturates, gaseous anaesthetics, steroids, narcotic analgesics, anticonvulsants and some antibiotics, cross the barrier by simple diffusion quite rapidly. This shows that the placental barrier is not as effective a barrier as BBB.
- Nutrients essential for the foetal growth are transported by carrier-mediated processes. Immunoglobulins are transported by endocytosis.
Fig. 43.Placental barrier and blood flow across it.
- Blood-Testis Barrier:
This barrier is located not at the capillary endothelium level but at sertoli-sertoli cell junction. It is the tight junctions between the neighbouring sertoli cells that act as the blood-testis barrier. This barrier restricts the passage of drugs to spermatocytes and spermatids.
Fig. 44. Blood-testis barrier.
2. ORGAN/TISSUE SIZE AND PERFUSION RATE
In the distribution phase of pharmacokinetics, the rate at which a drug reaches a specific tissue depends heavily on two physiological factors: the size of the organ and the perfusion rate (blood flow). Together, these determine the “capacity” of the tissue to take up the drug and the “speed” at which it arrives.
- Perfusion Rate (Blood Flow)
- Perfusion rate is defined as the volume of blood that flows per unit time per unit volume of the tissue. It is expressed in ml/min/ml of the tissue. The perfusion rate of various tissues is given in Below Table.
- Highly Perfused Organs: Organs like the liver, kidney, heart, and brain receive a massive percentage of cardiac output. In these tissues, the distribution of a drug is incredibly rapid.
- Poorly Perfused Organs: Tissues like fat (adipose), muscle, and bone have low blood flow. Consequently, it takes much longer (hours or even days) for a drug to reach therapeutic concentrations in these areas.
- Rate-Limiting Step: For most lipophilic drugs, the blood flow itself is the “bottleneck.” This is known as perfusion-limited distribution.
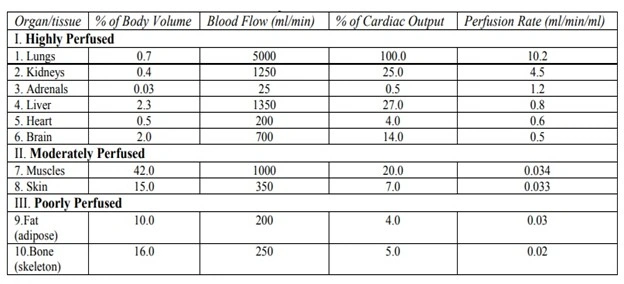
- Organ/Tissue Size
- The physical mass or volume of the tissue dictates the total capacity of that “compartment” to hold the drug. Larger the organ, more the capacity and vice versa.
- Large Organs (e.g., Muscle): Even though muscle has a moderate perfusion rate, its massive total body mass means it can act as a giant reservoir for certain drugs.
- Small Organs (e.g., Adrenals): An organ might be highly perfused, but if it is tiny, it will saturate with the drug almost instantly, and its impact on the overall plasma concentration of the drug will be minimal.
3. BINDING OF DRUGS TO TISSUE COMPONENTS
A drug in the body can bind to several components such as the plasma proteins, blood cells and haemoglobin (i.e. blood components) and extravascular proteins and other tissues. This topic is dealt comprehensively in chapter on Protein Binding of Drugs.
4. MISCELLANEOUS FACTORS AFFECTING DRUG DISTRIBUTION
- Age
Age is a profound physiological factor that alters drug distribution by changing body composition and organ function. The impact of age is most significant at the two extremes of life: neonates (newborns) and the elderly.- Neonates and Infants (Pediatrics)
In newborns, the body is physically and chemically different from an adult, which dramatically changes the Volume of Distribution (Vd).- Total Body Water (TBW): Neonates have a much higher percentage of body water (approx. 70–75%) compared to adults (60%).
- Impact: Water-soluble drugs (e.g., Gentamicin) distribute more widely, often requiring higher mg/kg doses to achieve therapeutic plasma levels.
- Body Fat: Newborns have significantly lower fat stores.
- Impact: Lipid-soluble drugs have a smaller volume of distribution.
- Plasma Protein Binding: Neonates have lower levels of albumin and reduced binding capacity.
- Impact: This leads to a higher free fraction of drugs higher distribution.
- Blood-Brain Barrier (BBB): The BBB is not fully developed (more permeable) in infants.
- Impact: Drugs that normally don’t enter the adult brain (like certain antibiotics or toxins) can easily reach the central nervous system in neonates.
- Total Body Water (TBW): Neonates have a much higher percentage of body water (approx. 70–75%) compared to adults (60%).
- The Elderly (Geriatrics)
- Increased Body Fat: With age, lean muscle mass decreases and body fat increases.
- Impact: Lipid-soluble drugs (e.g., Diazepam, Lidocaine) have an increased volume of distribution. They stay in the body much longer because they are stored in the expanded fat reservoirs.
- Decreased Total Body Water: The elderly have less intracellular and extracellular water.
- Impact: Water-soluble drugs (e.g., Digoxin, Ethanol) have a decreased volume of distribution, leading to higher, potentially toxic plasma concentrations from “standard” doses.
- Reduced Plasma Albumin: Albumin levels often decline due to age or poor nutrition.
- Impact: Increased free concentration of highly protein-bound drugs (e.g., Warfarin, Phenytoin), increasing the risk of adverse effects.
- Reduced Cardiac Output: Aging leads to a decrease in the perfusion rate to major organs.
- Impact: Slower distribution of drugs to the liver and kidneys, often slowing down the overall clearance.
- Increased Body Fat: With age, lean muscle mass decreases and body fat increases.
- Neonates and Infants (Pediatrics)
- Pregnancy
During pregnancy, the growth of uterus, placenta and foetus increases the volume available for distribution of drugs. The foetus represents a separate compartment in which a drug can distribute. The plasma and the ECF volume also increase but there is a fall in albumin content.
| Factor | Change in Pregnancy | Impact on Drug Distribution |
| Plasma Volume | ↑ | Dilutes water-soluble drugs (Lower Peak Conc.) |
| Body Fat | ↑ | Increases reservoir for lipid-soluble drugs |
| Plasma Albumin | ↓ | Increases “Free” (Active) drug fraction |
| Cardiac Output | ↑ | Increases rate of distribution to major organs |
- Obesity – The “Lipid Sink”
Obesity fundamentally shifts the ratio of fat to water in the body. Lipophilic Drugs (e.g., Diazepam, Thiopental): These have a massive affinity for adipose tissue. In obese patients, the Volume of Distribution increases significantly. - Diet
Dietary intake alters distribution primarily by changing blood chemistry and blood flow.- High-Fat Diets: Increase the levels of Free Fatty Acids (FFA) in the plasma. FFAs compete with acidic drugs (like Warfarin or Phenytoin) for binding sites on Albumin.
- Malnutrition/Low Protein: Leads to Hypoalbuminemia (low albumin). Result: Less “bus seats” for the drug to travel on. More free drug is available to diffuse into tissues, which can lead to an exaggerated pharmacological response.
- Hydration Status: Dehydration reduces the volume of extracellular fluid, decreasing the volume of distribution for water-soluble drugs and raising their plasma concentration.
- Disease States
A number of mechanisms may be involved in the alteration of drug distribution characteristics in disease states:- Cardiovascular Disease
- Reduced Perfusion: The heart can’t pump blood efficiently to peripheral tissues (muscle, fat, skin). The Impact: Distribution to these tissues is delayed. However, blood flow to the “core” (brain/heart) is often preserved, meaning the drug stays in the central compartment longer, potentially causing CNS toxicity.
- Liver Disease (Cirrhosis)
- Reduced Protein Synthesis: The liver stops making enough Albumin. The impact: Fluid accumulation in the abdomen creates a “third space.” Water-soluble drugs can get trapped in this peritoneal fluid, massively increasing their volume of distribution.
- Kidney Disease (Renal Failure)
- Reduces in the drug metabolism and increase in drug concentration in blood, hence increase the drug distribution.
- Cardiovascular Disease
VOLUME OF DISTRIBUTION / APPARENT VOLUME OF DISTRIBUTION
- A drug in circulation distributes to various organs and tissues. When the process of distribution is complete, different organs and tissues contain varying concentrations of drug which can be determined by the volume of tissues in which the drug is present. Since different tissues have different concentrations of drug, the volume of distribution cannot have a true physiologic meaning.
- However, there exists a constant relationship between the concentration of drug in plasma, C, and the amount of drug in the body, X.
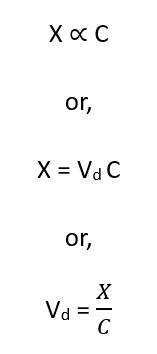
- Where,
- Vd = proportionality constant having the unit of volume (i.e., Liters/mL) and popularly called as apparent volume of distribution.
- It is defined as the hypothetical volume of body fluid into which a drug is dissolved or distributed. It is called asapparent volumebecause all parts ofthe body equilibrated with the drug do not have equal concentration.
- Thus, from equation Vd = X/C, Vd is given by the ratio:
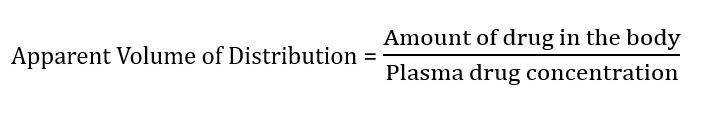
- Example No. 1.
Blood volume = 6 L
Amount of drug in the body (X) = 120 mg
Plasma drug concentration (C) = 20 mg/L
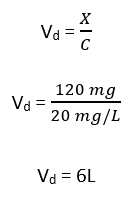
Which means, as the Vd is 6L and equal to the total blood volume, the drug has dissolved completely in the plasma but not distributed in the body.
- Example No. 2.
Blood volume = 6 L
Amount of drug in the body (X) = 120 mg
Plasma drug concentration (C) = 10 mg/L (some drug is distributed in organs, hence the concentration in the blood has reduced)
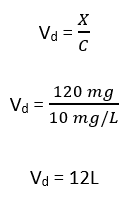
Which means, as the Vd is 12L and is more than the total blood volume, the drug has dissolved completely in the plasma and distributed in the body as well.
Vd – Total blood in the body = Volume of other body fluids i.e., 12 L – 6 L = 6 L, the drug has been distributed in 6 L of body fluid (ECF and ICF)
EXTRA – Knowledge purpose
- The apparent volume of distribution bears no direct relationship with the real volume of distribution.
- The real volume of distribution has direct physiologic meaning and is related to the body water. The body water is made up of 3 distinct compartments as shown in the below Table.
Fluid Compartments of a 70 Kg Adult
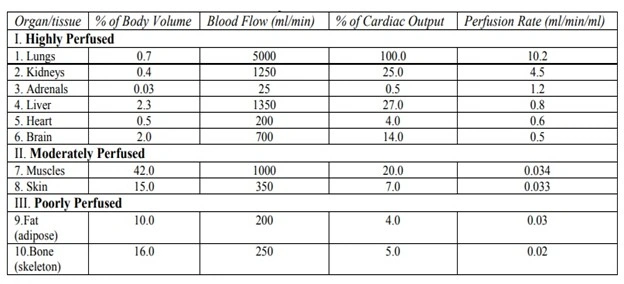
- The volume of each of these real physiologic compartments can be determined by use of specific tracers or markers (given in below Table).
- The plasma volume can be determined by use of substances of high molecular weight or substances that are totally bound to plasma albumin, for e.g. high molecular weight dyes such as Evans blue, indocyanine green and I-131 albumin. When given i.v., these remain confined to the plasma. The total blood volume can also be determined if the haematocrit is known.The extracellular fluid (ECF) volume can be determined by substances that easily penetrates the capillary membrane and rapidly distribute throughout the ECF but do not cross the cell membranes, for e.g. the Na+, Cl –, Br–, SCN– and SO42– ions and inulin, mannitol and raffinose. However, none of these substances are completely kept out of the cells. The ECF volume, excluding plasma is approximately 15 litres.
- The total body water (TBW) volume can be determined by use of substances that distribute equally in all water compartments of the body (both intra- and extracellular), for e.g. heavy water (D2O), tritiated water (HTO) and lipid soluble substances such as antipyrine. The intracellular fluid volume is determined as the difference between the TBW and ECF volume. The intracellular fluid volume including those of blood cells is approximately 27 litres.
Markers Used to Measure the Volume of Real Physiological Compartments
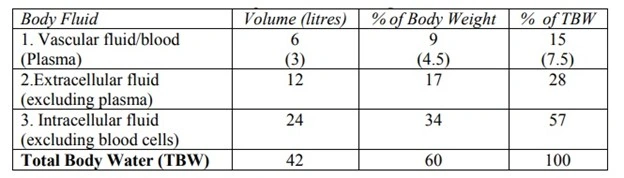
Protein Binding of Drugs (Binding of drugs)
- A drug in the body can interact with several tissue components of which the two major categories are –
- Blood, and
- Extravascular tissues.
- The interacting molecules are generally the macromolecules such as proteins, DNA or adipose.
- The proteins are particularly responsible for such an interaction. The phenomenon of complex formation with proteins is called as protein binding of drugs.
Protein binding may be divided into –
- Intracellular binding –where the drug is bound to a cell protein which may be the drug receptor; if so, binding elicits a pharmacological response. These receptors with which drug interact to show response are called as primary receptors.
- Extracellular binding –where the drug binds to an extracellular protein but thebinding does not usually elicit a pharmacological response. These receptors are called secondary or silent receptors.
- The most important extracellular proteins or silent receptors are plasma proteins, in particular albumin. Binding to such proteins is important from the viewpoint that the bound drug is both pharmacokinetically as well as pharmacodynamically inert i.e. an extracellular protein bound drug is neither metabolised nor excreted nor it is active pharmacologically.
- A bound drug is also restricted since it remains confined to a particular tissue for which it has greater affinity. Moreover, such a bound drug, because of its enormous size, cannot undergo membrane transport and thus its half-life is increased.
Mechanisms of Protein-Drug Binding
- Binding of drugs to proteins is generally reversible which suggests that it generally involves weak chemical bonds such as –
- Hydrogen bonds
- Hydrophobic bonds
- Ionic bonds, or
- van der Waal’s forces.
- Irreversible drug binding, though rare, arises as a result of covalent binding and is often a reason for the carcinogenicity or tissue toxicity of the drug; for example, covalent binding of chloroform and paracetamol metabolites to liver results in hepatotoxicity.
Binding of drugs falls into 2 classes:
- Binding of drugs to blood components like—
- Plasma proteins
- Blood cells
- Binding of drugs to extravascular tissue proteins, fats, bones, etc.
- The influence of binding on drug disposition and clinical response is shown in Fig. 45.
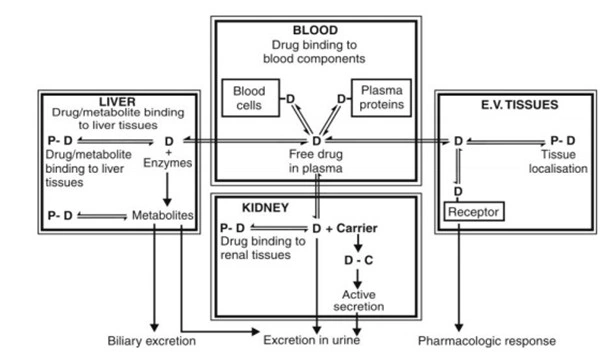
Fig. 45. Protein-drug binding: Binding of drugs to various tissue components and its influence on disposition and clinical response. Note that only the unbound drug moves reversibly between the compartments.
- Of all types of binding, the plasma protein-drug binding is the most significant and most widely studied.
PLASMA PROTEIN-DRUG BINDING
- Following entry of a drug into the systemic circulation, the first things with which it can interact are blood components like plasma proteins, blood cells and haemoglobin (see below Table).
- The main interaction of drug in the blood compartment is with the plasma proteins which are present in abundant amounts and in large variety. The binding of drugs to plasma proteins is reversible. The extent or order of binding of drugs to various plasma proteins is:
Albumin > α1 Acid Glycoprotein > Lipoproteins > Globulins. - Blood Proteins to which Drugs Bind
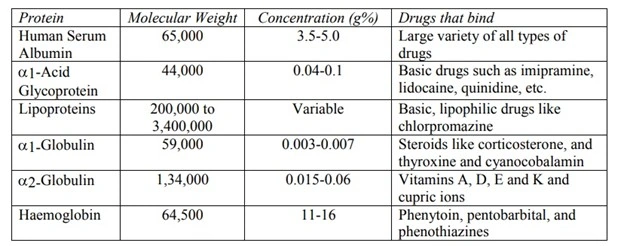
Binding of Drugs to Human Serum Albumin
- The human serum albumin (HSA), having a molecular weight of 65,000, is the most abundant plasma protein (59% of total plasma and 3.5 to 5.0 g%) with a large drug binding capacity.
- The therapeutic doses of most drugs are relatively much smaller and their plasma concentration do not normally reach equimolar concentration with HSA.
- The HSA can bind several compounds having varied structures.
- A large variety of drugs ranging from weak acids, neutral compounds to weak bases bind to HSA. Four different sites on HSA have been identified for drug-binding (Fig. 46). They are:
- Site I: Also called as warfarin and azapropazone binding site, it represents the regionto which large number of drugs are bound, e.g. several NSAIDs, sulphonamides, phenytoin, sodium valproate and bilirubin.
- Site II: It is also called as the diazepam binding site. Drugs which bind to this region include benzodiazepines, medium chain fatty acids, ibuprofen, ketoprofen, tryptophan, cloxacillin, probenicid, etc.
- Site I and site II are responsible for the binding of most drugs.
- Site III: is also called as digitoxin binding site.
- Site IV: is also called as tamoxifen binding site.
- Very few drugs bind to sites III and IV.
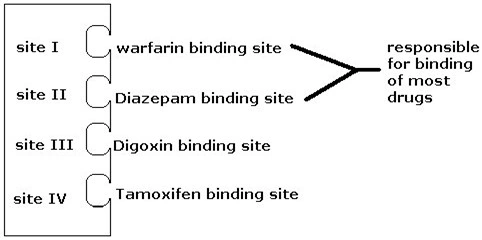
Fig. 46. Four major binding sites on Human serum albumin
Binding of Drugs to α1-Acid Glycoprotein (α1-AGP or AAG)
- It has a molecular weight of 44,000 and a plasma concentration range of 0.04 to 0.1 g%.
- It binds to a number of basic drugs like imipramine, amitriptyline, nortriptyline, lidocaine, propranolol, quinidine and disopyramide.
Binding of Drugs to Lipoproteins
- Lipoproteins serve as the primary transport system for highly lipophilic (fat-soluble) and basic drugs.
- The molecular weight of lipoproteins varies from 2 lakhs to 34 lakhs depending on their chemical composition.
Binding of Drugs to Globulins
- Several plasma globulins have been identified and are labelled as α, β and γ- globulins.
- It binds number of steroidal drugs, vitamins A, D, E and K.
TISSUE BINDING OF DRUGS (TISSUE LOCALIZATION OF DRUGS)
- Distribution of drug results in entry of drug at tissue level. On distribution drug binds with several tissue components. Tissue binding of drug affects its apparent volume of distribution.
- The increase in tissue binding increases apparent volume of distribution. Apparent volume of distribution is the ratio of amount of drug in the body to the plasma concentration of unbound drug. The increase in tissue binding results in decrease in plasma concentration of drug due to localization of drug at tissue level, which eventually increase apparent volume of distribution.
- In addition to this, tissue binding of drug results in increase in its biological half. However certain drugs or their metabolites irreversibly bind to tissues results in precipitation of toxicity. For example, metabolites of paracetamol bind irreversibly to hepatic tissues and produce toxicity.
- For majority of drugs that bind to extravascular tissues, the order of binding is:
Liver > Kidney > Lung > Muscles - The common examples of tissue drug binding are:
- Liver: Meatbolites of paracetamol and carbon tetrachloride bind irreversibly to liver tissues resulting in hepatic injury.
- Lungs: Weak basic drugs like imipramine, chlorpromazine and antihistamines bind with lung tissues results in accumulate of these drugs in lungs.
- Kidneys: Certain heavy metals like lead, mercury and cadmium bind with protein present in kidneys results in renal toxicity.
- Skin: Drugs like chloroquine and phenothiazines bind with skin pigment melanin.
- Eyes: Binding of chloroquine and phenothiazines to retinal pigment results in retinopathy.
- Bones: Tetracycline binds to bones and teeth. Administration of drug to infants or children results in permanent brown-yellow discoloration of teeth.
- Fats: Certain drugs like thiopental bind to adipose tissues and accumulate in tissues.
Comparison Between Plasma Protein-Drug Binding and Tissue-Drug Binding
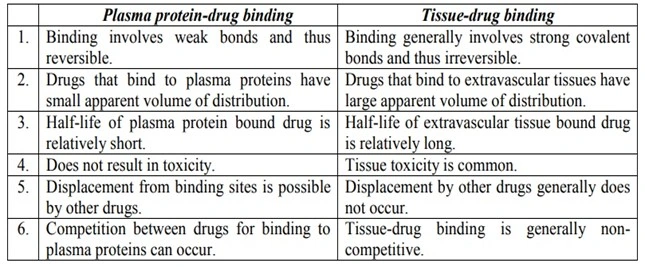
FACTORS AFFECTING PROTEIN-DRUG BINDING
Factors affecting protein-drug binding can be broadly categorized as—
- Drug related factors
- Physicochemical characteristics of the drug
- Concentration of drug in the body
- Affinity of a drug for a particular binding component
- Protein/tissue related factors
- Physicochemical characteristics of the protein or binding agent
- Concentration of protein or binding component
- Number of binding sites on the binding agent
- Drug interactions
- Competition between drugs for the binding site (displacement interactions)
- Competition between the drug and normal body constituents
- Allosteric changes in protein molecules
- Patient-related factors
- Age
- Intersubject variations
- Disease states
Factors affecting Protein-Drug Binding Explanation
1. DRUG RELATED FACTORS
a. Physicochemical Characteristics of the Drug
- Lipophilicity: This is the most significant factor. Most protein binding sites have lipophilic pockets. Hence, more the lipophilicity of drug, more the binding.
Lipophilicity ∝ protein binding - Ionization and pKa: The ionisation of drug determines which protein it targets:
- Acidic Drugs: Usually exist as anions. They have a high affinity for Human Serum Albumin (HSA).
- Basic Drugs: Usually exist as cations. They prefer binding to alpha-1-Acid Glycoprotein (AAG) or Lipoproteins.
- Highly ionized drugs tend to have lower protein binding because they are comfortable staying dissolved in the water of the plasma.
b. Concentration of Drug in the Body
- The extent of protein-drug binding can change with both changes in drug as well as protein concentration.
- The concentration of drugs that bind to HSA (Human serum albumin) does not have much of an influence, as the therapeutic concentration of any drug is insufficient to saturate it.
- However, therapeutic concentration of lidocaine can saturate AAG (Alpha1 acid glycoprotein) with which it binds as the concentration of AAG is much less in comparison to that of HSA in blood. In such condition, after saturation, the drug will be in the free form.
c. Drug-Protein/Tissue Affinity
- All types of drugs have affinity towards Human serum albumin.
- Basic drugs have affinity towards Alpha1 acid Glycoprotein.
- Lipophilic drugs have affinity towards Lipoproteins.
- Steroids and Vitamins have affinity towards Globulins
- Digoxin has more affinity for proteins of cardiac muscles than those of skeletal muscles or plasma.
PROTEIN/TISSUE RELATED FACTORS
a. Physicochemical Properties of Protein/Binding Component
- Lipoproteins and adipose tissue tend to bind lipophilic drugs by dissolving them in their lipid core.
- Globulins have an affinity towards steroids and vitamins.
b. Concentration of Protein/Binding Component
- Among the plasma proteins, binding predominantly occurs with albumin, as it is present in a higher concentration in comparison to other plasma proteins.
- The amount of several proteins and tissue components available for binding, changes during disease states.
- When there is a reduction in the concentration of a particular protein for binding, the drug searches for a different protein for binding.
c. Number of Binding Sites on the Protein
- More Sites = Higher Capacity: If a protein molecule like Albumin has multiple binding sites (it has 4 distinct pockets), it can carry a much higher load of the drug before reaching saturation.
- Independent vs. Identical Sites: In many cases, these sites are “independent,” meaning a drug binding to Site 1 doesn’t make it harder or easier for another drug molecule to bind to Site 2.
3. DRUG INTERACTIONS
a. Competition Between Drugs for the Binding Sites (Displacement Interactions)
- When two or more drugs can bind to the same site, competition between them for interaction with the binding site results. If one of the drugs (drug A) is bound to such a site, then administration of another drug (drug B) having affinity for the same site results in displacement of drug A from its binding site. Such a drug-drug interaction for the common binding site is called as displacement interaction.
- The drug A here is called as the displaced drug and drug B as the displacer.
- Ex.: Warfarin and phenylbutazone have affinity for HSA. Administration of phenylbutazone to a patient on warfarin therapy results in displacement of warfarin from its binding site. The free warfarin may cause adverse hemorrhagic reactions which may be lethal.
- Displacement interactions can result in unexpected rises in free concentration of the displaced drug which may enhance clinical response or toxicity.
b. Competition Between Drugs and Normal Body Constituents
- Displacement of drug by Normal Body Constituents: During fasting, stress, or certain hormonal changes, Free Fatty Acids levels rise. FFAs compete with acidic drugs for binding with albumin. This can lead to an increase in the free fraction of drugs like diazepam or warfarin and leads to drug toxicity.
d. Allosteric Changes in Protein Molecule
- The process involves alteration of the protein structure by the drug or its metabolite thereby modifying its binding capacity.
- The agent that produces such an effect is called as allosteric effector.
- Some agents produce positive effects, i.e., increase the binding capacity of protein and some agents produce negative effects, i.e., decrease the binding capacity of protein.
4. PATIENT-RELATED FACTORS
a. Age
- Protein-drug binding differs due to protein content in various age groups.
- Neonates: Albumin content is low in newborn; as a result, the unbound concentration of drug that primarily binds to albumin is increased.
- Elderly: In old age, the albumin content is lowered and free concentration of drugs that bind primarily to it is increased. Old age is also characterized by an increase in the levels of AAG and thus decreased free concentration is observed for drugs that bind to it.
b. Intersubject variations:
- Related to the genetic, some individuals have slight structural variations in their albumin or AAG molecules due to their DNA. This can change the “shape” of the binding pocket, making the drug fit more loosely or tightly.
c. Disease States:
- Several pathological conditions are associated with alteration in protein content. Since albumin is the major drug binding protein, hypoalbuminaemia can severely impair protein-drug binding.
- Hypoalbuminaemia is caused by several conditions like aging, CCF, trauma, burns, inflammatory states, renal and hepatic disorders, pregnancy, surgery, cancer, etc. Almost every serious chronic illness is characterized by decreased albumin content. Some of the diseases that modify protein-drug binding are depicted in below Table
- Influence of Disease States on Protein-Drug Binding
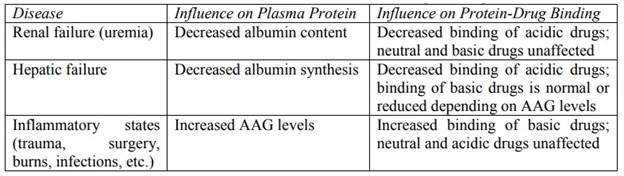
KINETICS OF PROTEIN-DRUG BINDING
1. If P represents proteins and D the drug, then applying law of mass action to reversible protein-drug binding, we can write:
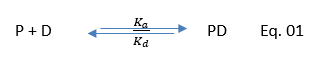
At equilibrium,

where,
[P] = concentration of free protein
[D] = concentration of free drug
[PD] = concentration of protein-drug complex
Ka = association rate constant
Kd = dissociation rate constant Ka > Kd indicates forward reaction i.e. protein-drug binding is favoured.
2. If PT is the total concentration of protein present, bound and unbound, then:

If r is the number of moles of drug bound to total moles of protein, then,
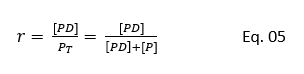
Substituting the value of [PD] from Eq. 03 in Eq.05 we get:
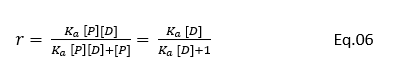
Eq. 06 holds when there is only one binding site on the protein and the protein-drug complex is a 1:1 complex. If more than one or N number of binding sites are available per mole of the protein, then:

The value of association constant, Ka and the number of binding sites N can be obtained by plotting Eq. 07 in four different ways as shown below (see Fig. 47).
- Direct Plot
- Scatchard Plot
- Klotz Plot
- Hitchcock Plot
Explain anyone.
Direct Plot:
Direct Plot is made by plotting r versus [D] as shown in Fig. 47a. Note that when all the binding sites are occupied by the drug, the protein is saturated and plateau is reached. At the plateau, r = N. When r = N/2, [D] = 1/Ka.
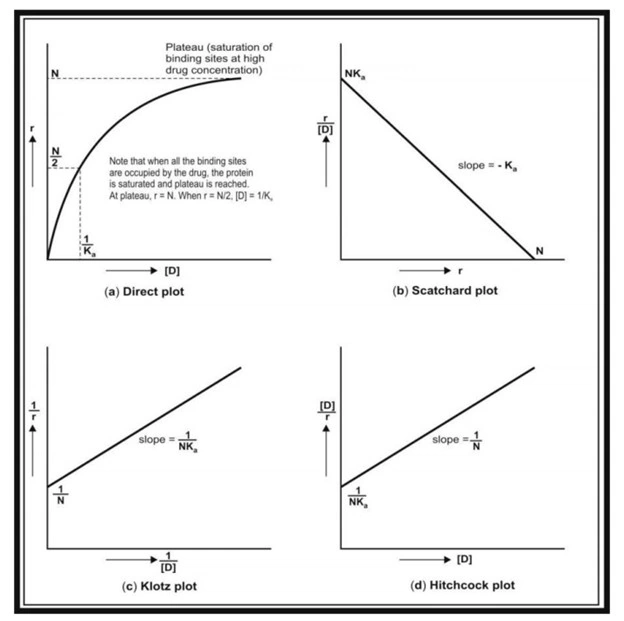
Fig. 47. Plots used for the study of protein-drug binding. (a) Direct plot; (b) Scatchard plot; (c) Klotz plot; and (d) Hitchcock plot.
Clinical significance of protein binding of drugs
- Absorption: The absorption equilibrium is attained by transfer of free drug from the site of administration into the systemic circulation and when the concentration in these two compartments become equal. Following equilibrium, the process may stop. However, binding of the absorbed drug to plasma proteins decreases free drug concentration and disturbs such equilibrium. Thus, sink conditions and the concentration gradient are re-established which now act as the driving force for further absorption.
- Systemic Solubility of Drugs: Protein-drug complex can be used as a vehicle in drug distribution.
- Distribution: Plasma protein binding restricts the entry of drugs that have specific affinity for certain tissues. This prevents accumulation of a large fraction of drug in such tissues and thus, subsequent toxic reactions. Plasma protein-drug binding thus favours uniform distribution of drugs throughout the body by its buffer function (maintains equilibrium between the free and the bound drug).
- Tissue Binding, Apparent Volume of Distribution and Drug Storage: A drug that is extensively bound to blood components remains confined to blood. Such a drug has a small volume of distribution. A drug that shows extravascular tissue binding has a large volume of distribution. A tissue or blood component that has great affinity for a particular drug act as a depot or storage site for that drug.
- Elimination: Only the unbound or free drug is capable of being eliminated. This is because the drug-protein complex cannot penetrate into the metabolising organ (liver). The large molecular size of the complex also prevents it from getting filtered through the glomerulus. Thus, drugs which are more than 95% bound are eliminated slowly i.e. they have long elimination half-lives; for example, tetracycline, which is only 65% bound, has an elimination half-life of 8.5 hours in comparison to 15.1 hours of doxycycline which is 93% bound to plasma proteins.
- Displacement Interactions and Toxicity: As stated earlier, displacement interactions are significant in case of drugs which are more than 95% bound. This is explained from the example given in below Table. A displacement of just 1% of a 99% bound drug results in doubling of the free drug concentration i.e. a 100% rise. For a drug that is bound to a lesser extent e.g. 90%, displacement of 1% results in only a 10% rise in free drug concentration which may be insignificant clinically.
Influence of Percent Binding and Displacement on Change in Free Concentration of Drugs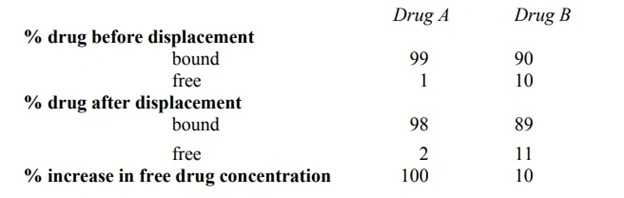
- Diagnosis: The chlorine atom of chloroquine when replaced with radiolabelled I-131 can be used to visualize melanomas of the eye since chloroquine has a tendency to interact with the melanin of eyes. The thyroid gland has great affinity for iodine containing compounds; hence any disorder of the same can be detected by tagging such a compound with a radioisotope of iodine.
- Therapy and Drug Targeting: The binding of drugs to lipoproteins can be used for site-specific delivery of hydrophilic drugs. This is particularly useful in cancer therapies since certain tumour cells have greater affinity for LDL than normal tissues.
Questions.
10 Marks.
- Write a note on absorption and mechanism of drug absorption.
- Classify factors influencing absorption of drugs. Explain physicochemical factors in detail.
- Explain the pharmaceutical factors and biological or patient related factors influencing drug absorption through GIT.
- Define biopharmaceutics. Discuss in detail about kinetics of protein binding.
5 Marks.
- Enlist various mechanisms of drug absorption through GIT. Explain active transport and passive diffusion/ transport.
- Compare and contrast active transport and passive transport/ diffusion.
- Explain apparent volume of distribution.
- Enumerate the factores affecting protein drug binding. Explain any two factors.
- Explain tissue permeability of drug in detail.
- Explain kinetics of protein binding.
- Write about clinical significance of protein binding.
2 Marks.
- Define absorption and distribution of drug.
- Pore transport in drug absorption.
- What are pinocytosis and phagocytosis?
- Enlist physicochemical factors affecting drug absorption.
- What is the effect of food on absorption of drugs.
- What is polymorphism?
- Define protein binding.
- Enlist the factors affecting protein binding.
- List out the proteins responsible protein binding.
- Define apparent volume of distribution and write its significance.