
Unit IV – Parenteral Products (10 hrs.)
Syllabus:
Parenteral Products:
- Definition, types, advantages and limitations. Preformulation factors and essential requirements – vehicles, additives, importance of isotonicity.
- Production procedure, production facilities and controls, aseptic processing.
- Formulation of injections, sterile powders, large volume parenterals and lyophilized products.
- Containers and closures selection, filling and sealing of ampoules, vials and infusion fluids. Quality control tests of parenteral products.
Ophthalmic Preparations:
- Introduction, formulation considerations; formulation of eye drops, eye ointments and eye lotions; methods of preparation; labeling, containers; evaluation of ophthalmic preparations.
Parenteral Products:
- The term Parenteral has been derived from the Greek word Para and enteron, which means outside the intestine.
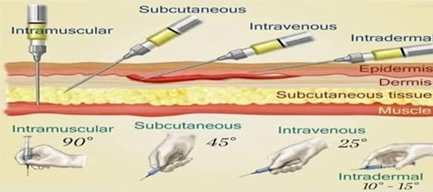
- These are unique dosage forms as they are administered by injecting directly into the body tissues through skin and mucous membranes.
Definition:
Parenteral products are sterile preparations containing one or more active ingredients intended for administration by injection, infusion or implantation into the body. They are packaged in either single-dose or multi dose containers.
Types of Parenteral Products:
Parenteral products are sterile preparations administered by injection, infusion, or implantation.
They are classified based on:
- Based on Dosage Form.
- Based on Volume.
- Special Parenteral Products.
1. Based on Dosage Form
a) Aqueous Solutions
- Examples: NS, dextrose, adrenaline injection
- Water-based, clear liquids
b) Non-aqueous Solutions
- Examples: Vitamin A injection, progesterone injection
- Use oil vehicles (sesame oil, arachis oil)

c) Suspensions
- Insoluble drug particles dispersed in a liquid
- Used for depot effect
- Examples: Penicillin G procaine, insulin suspension
d) Emulsions
- Mixtures of oil and water
- Examples: IV lipid emulsions, propofol
e) Dry Powders for Reconstitution
- Sterile powders to be mixed before injection
- Examples: Ceftriaxone, amoxicillin-clavulanic acid

f) Lyophilized (Freeze-Dried) Products
- Special type of dry powder for stability
- Examples: Anti-cancer drugs, monoclonal antibodies
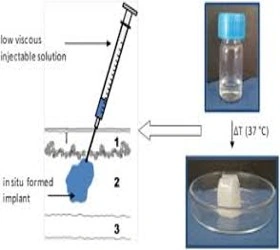
g) Implants
- Examples: contraceptive implants (etonogestrel)
- Long-acting drug delivery systems
2. Based on Volume
a) Small Volume Parenterals (SVPs)
- Volume ≤ 100 mL
- Used for injections and small infusions
- Examples: 1 mL insulin, 10 mL B-complex
b) Large Volume Parenterals (LVPs)
- Volume > 100 mL
- Used for IV fluids and nutrition
- Examples: 500 mL NS (Normal saline), 1 L RL (Ringer’s Lactate), TPN (Total Parenteral Nutrition)

3. Special Parenteral Products
a) Radiopharmaceutical Injections
- Used for imaging and therapy
- Example: Technetium-99m agents
b) Vaccines & Biologics
- Include antigens, antibodies, proteins
- Example: vaccines, insulin, mAbs (Monoclonal Antibodies)
c) Irrigation Solutions
- Example: glycine irrigation, sterile water irrigation
- Sterile solutions used during surgery

Advantages of Parenteral Products:
- Rapid Onset of Action
- Drugs reach systemic circulation directly.
- Ideal for emergencies (shock, cardiac arrest, anaphylaxis).
- 100% Bioavailability
- No absorption barriers.
- Accurate and predictable dose reaches the bloodstream.
- Useful for Unconscious or Uncooperative Patients
- It can be given to patients who cannot swallow or are vomiting.
- Suitable for Drugs Not Absorbed Orally
- Proteins, peptides, insulin, vaccines, and drugs destroyed by GIT enzymes.
- Bypasses First-Pass Metabolism
- Avoids hepatic metabolism that occurs with oral drugs.
- Precise Control Over Drug Levels
- IV infusion allows controlled, steady plasma concentration.
- Long-Acting (Depot) Formulations Possible
- IM injections and implants provide sustained release for days to months.
- Suitable for Irritating or Tissue-Damaging Drugs
- Some drugs cannot be given orally due to irritation or inactivation.
- Immediate Correction of Fluid & Electrolyte Imbalance
- IV fluids (NS, RL, dextrose) quickly restore hydration and electrolytes.
Limitations of Parenteral Products:
- Pain, Discomfort & Needle Phobia
- Injections can cause pain, irritation, and anxiety.
- Risk of Infection
- Breaks skin barrier → chance of contamination.
- It requires strict sterility and aseptic technique.
- Requires Skilled Personnel
- Proper administration needs trained staff.
- Errors (IV bolus, wrong route) can be dangerous.
- Higher Cost
- Manufacturing, packaging, and handling are expensive due to sterility requirements.
- Difficult to Reverse
- Once injected, the drug cannot be withdrawn.
- Overdose or adverse effects are harder to manage.
- The administration of drug through wrong route of injection may prove to be fatal.
- Risk of Tissue Damage
- Poor technique can cause nerve damage, abscess, thrombophlebitis, and necrosis.
- Stability Issues
- Some formulations (solutions/suspensions) have limited shelf life.
- Sensitive to temperature, pH, light, particulate matter.
- Limited Self-Administration
- Most injections cannot be given by the patient themselves (except insulin, epipens).
- It is difficult to save a patient when overdose is given.
- There are chances of sensitivity reaction or allergic reaction of a drug by an individual. These reactions are sometimes very fatal and lead to death.
Preformulation Factors:
Definition:
Preformulation factors are the physicochemical, biopharmaceutical, and mechanical properties of a drug substance that influence the selection of a suitable dosage form and formulation strategy.
Or
Preformulation of parenterals involves systematic studies carried out before formulation development to ensure that the drug is safe, stable, sterile, and effective when administered by injection.
The following are the factors:
- Physicochemical Factors
- Biopharmaceutical Factors
- Mechanical / Processing Factors
- Stability related Factors
1. Physicochemical Factors
a) Physical Properties
- Polymorphism: Different crystal forms show different solubility, melting point, stability.
- Particle Size & Shape: Affects dissolution rate, flowability, bioavailability.
- Hygroscopicity: Tendency to absorb moisture.
- Density (true, bulk) & Compressibility: Important for tablet and capsule design.
- Flow Properties: Angle of repose, Carr’s index, Hausner ratio.
- Crystalline vs Amorphous Nature: Amorphous forms are more soluble but less stable.
b) Chemical Properties
- Solubility (aqueous & non-aqueous): Determines dissolution and absorption.
- pKa & pH–Solubility Profile: Helps in salt selection and formulation pH.
- (Partition Coefficient log P): Indicates lipid solubility and permeability. It Measures how a chemical distributes between two immiscible liquids (like oil and water) at equilibrium.
- Chemical Stability: Hydrolysis, oxidation, photolysis, racemization.
- Drug–Excipient Compatibility: Checked by DSC (Differential Scanning Calorimetry), IR (Infrared Spectroscopy), XRD (X-ray Diffraction) to avoid interactions.
2. Biopharmaceutical Factors
- Absorption characteristics: Describe the rate and extent at which a drug reaches systemic circulation after non-intravenous parenteral administration (IM, SC, intradermal).
- Permeability (BCS classification): Permeability refers to the ability of a drug to cross biological membranes and reach systemic circulation or target tissues. In preformulation, it helps predict distribution, onset of action, and tissue penetration, even though parenterals bypass the GI tract.
- Bioavailability: Bioavailability is the rate and extent to which the active drug reaches systemic circulation in an unchanged form after administration.
- First-pass metabolism tendencies.
3. Mechanical / Processing Factors
- Compressibility: Compressibility is the ability of a powder or granules to decrease in volume under applied pressure and form a coherent compact.
Helps in Lyophilized (freeze-dried) injections, Implants and depot systems and Dry powder for reconstitution - Flowability: Flowability refers to the ability of a powder or granular material to flow smoothly and consistently under a given set of conditions.
- Wettability: Wettability is the ability of a solid surface (especially a powder) to be penetrated or spread over by a liquid.
- Lubrication Sensitivity: Interaction with magnesium stearate, etc.
4. Stability-Related Factors
Stability is the ability of a drug substance or formulation to retain its physical, chemical, and biological properties when exposed to heat/light/pH/moisture.
- Thermal stability: It is the ability of a substance to resist chemical or physical changes—such as decomposition, deformation, or loss of properties—when exposed to elevated temperatures.
- Photostability: It is the ability of a substance to resist chemical or physical changes—such as decomposition, deformation, or loss of properties—when exposed to light.
- pH stability: pH stability refers to the ability of a substance, solution, or biological system to maintain its chemical and physical integrity, functional activity, or consistent pH level when exposed to acidic or basic conditions.
- Moisture sensitivity: It is the degree to which a material or substance undergoes physical or chemical changes when exposed to water vapor in the environment
- Shelf-life prediction (Arrhenius equation): Shelf-Life Prediction is the estimation of the time-period during which a drug product remains within acceptable limits of potency, safety, and quality under specified storage conditions.
Essential requirements for Formulation:
- The formulations of parenteral preparations need careful planning, thorough knowledge of medicaments and additives to be used.
- The excess use of additives in parenteral products should be avoided as some of these may interfere with the drug.
- In the preparation of parental products, the following substances are added to make a stable preparation.
- Vehicles –
a. Aqueous vehicles
b. Non–aqueous vehicles - Additives –
a. Solubilizing agents
b. Stabilizers
c. Buffering agents
d. Antibacterial agents
e. Chelating agents
f. Suspending, emulsifying and wetting agent
g. Tonicity factors
1. Vehicles:
There are two types of vehicles, which are commonly used for the preparation of injections. i.e.,
a. Aqueous vehicle and
b. Non-aqueous vehicle.
a. Aqueous vehicle –
- Water is used as vehicle for majority of injections because water is tolerated well by the body and is safest to administer.
- Water for injection is sterile water, which is free from volatile, non- volatile impurities and from pyrogens.
- Pyrogens are by-product of bacterial metabolism.
- Pyrogens are Lyposaccharide, thermostable, soluble in water, unaffected by bactericide and can pass through bacterial proof filters.
- Pyrogens can be removed from water by simple distillation process using an efficient trap which prevents the pyrogen to enter the condenser.
- Immediately after the preparation of water for injection, it is filled in to the final container, sealed and sterilized by autoclaving.
- Water for injection, contaminated with pyrogens may cause rise in body temperature if injected. Hence, test for pyrogen is done to ensure that water for injection is free from pyrogens.
The aqueous vehicle used are –
– Water for injection
– Water for injection free from CO2 (carbon dioxide)
– Water for injection free from dissolved air (oxygen).
b. Non-aqueous vehicles: –
- The commonly used non-aqueous vehicles are oils and alcohols.
- Fixed oil, such as arachis oil, cottonseed oil, almond oil and sesame oil are used as vehicle.
- Oily vehicles are generally used when a depot effect of drug is required, or the medicaments are insoluble or slightly soluble in water or the drug is soluble in oil example dimercaprol injection by using arachis oil as vehicle.
- Ethyl alcohol is used in the preparation of hydrocortisone injections. Hydrocortisone is insoluble in water, hence the solution is made in 50% alcohol.
- Alcohol causes pain and tissue damage at the site of injections. Therefore, it is not used commonly.
- Propylene glycol is used as a vehicle in the preparation of digoxin injections. It is relatively non-toxic, but it causes pain on S/C or I/M injection.
- Sometime polyethylene glycol and glycerin usually diluted with sterile water are used to prepare solutions for injections. They are used as solvent as well as to increase the stability of certain preparations.
2. Additives:
- These substances are added to increase the stability or quality of the product.
- The additives should be used only when it is necessary to use them.
- While selecting the additives, care must be taken that they should be compatible both physically and chemically with the entire formulation.
- They should be added in minimum possible quantity.
The following additives are commonly used in preparing stable parental preparations.
a. Solubilizing agents: –
- These are used to increase the solubility of drugs which are slightly soluble in water.
- The solubility of drug is increased by using surface active agent like tweens and polysorbate or by using co-solvents like alcohol, propylene glycol, PEG 300 or 400, sorbitol.
b. Stabilizers: –
- The drugs in the form of solution are more liable to deteriorate (become progressively worse) due to oxidation and hydrolysis.
- Stabilizers are added in the formulation to prevent oxidation and hydrolysis.
- The oxidation can be prevented by adding a suitable antioxidant such as, thiourea, ascorbic acid, sodium metabisulphite, or the product is sealed in an atmosphere of Nitrogen or Carbon dioxide.
- Hydrolysis can be prevented by using a non-aqueous vehicle or by adjusting the pH of the preparation.
- Examples of Antioxidants:
- Water soluble: Sulfurous acid salts, Ascorbic acid isomers, Thiol derivatives
- Oil soluble: Propyl gallate, Butylated hydroxyanisole, Ascorbyl palmitate, Alpha Tocopherol
c. Buffering agents: –
- The degradation of the preparation, which is due to change in pH, can be prevented by adding a suitable buffer to maintain the desired PH.
| pH | Buffer system | Concentration (%) |
| 3.5-5.7 | Acetic acid-acetate | 1-2 |
| 2.5-6.0 | Citric acid- citrate | 1-5 |
| 6.0-8.2 | Phosphoric acid- phosphate | 0.8-2 |
| 8.2-10.2 | Glutamic acid- glutamate | 1-2 |
d. Antibacterial agents: –
- These substances are added in adequate quantity to prevent the growth of microorganisms during storage. So, these substances act as preservatives.
- Parenteral products are sterilized by filtration method and / or terminal sterilization.
- Antibacterial agents in parenteral preparations are added to multidose injections to prevent microbial growth during storage and repeated use.
- Some typical preservatives used in parenteral suspensions and their commonly used concentrations are as follows. –
| Benzyl alcohol (0.9% to 1.5%) | Methylparaben (0.18% to 0.2%) |
| Propylparaben (0.02%) | Benzalkonium chloride (0.01% to 0.02%) |
| Thiomersal (0.001% to 0.01%) |
e. Chelating agent: –
- Chelating agents such as EDTA (Ethylene diamine Tetra acetic acid) and its salts, sodium or potassium salts of citric acid are added in the formulation to chelate the metallic ions present in the formulation.
- To enhance stability and safety by binding to trace metal ions.
- They form a Complex which gets dissolved in the solvent.
| S. No. | Additives | Concentration range (%) |
| 1 | EDTA disodium | 0.00368-0.05 |
| 2 | EDTA calcium disodium | 0.04 |
| 3 | EDTA tetrasodium | 0.01 |
f. Suspending, emulsifying and wetting agents:-
- The suspending agents are used to improve the viscosity and to suspend the particles for a long time. Methyl cellulose, carboxy-methyl cellulose, gelatin and acacia are commonly used as suspending agents.
- The emulsifying agents are used in sterile emulsions for this purpose is lecithin.
- The wetting agents are used to reduce the interfacial tension between the solid particles and the liquid, to prevent the formulation of lumps.
- They also act as antifoaming agents to subside the foam produced during manufacturing and shaking of the preparation.
- Examples of suspending agents, emulsifying agents and wetting agents.
| Category | Common Examples |
| Suspending Agents | Xanthan Gum, Methylcellulose (MC), Bentonite, Carbomer, and Tragacanth. |
| Emulsifying Agents | Lecithin, Polysorbate 80 (Tween 80), Glyceryl Monostearate, and Sodium Stearoyl Lactylate. |
| Wetting Agents | Sodium Lauryl Sulfate (SLS), Poloxamer 188, Glycerin, and Dioctyl Sodium Sulfosuccinate (Docusate). |
g. Tonicity factors: –
- Parenteral preparation should be isotonic with blood plasma or other body fluids.
- Solution having the same osmotic pressure as some other solution, especially one in blood, a cell or a body fluid.
- The isotonicity of the solution may be adjusted by adding sodium chloride, dextrose and boric acid etc. in suitable quantities.
- These substances should be compatible with other ingredients of the formulation.
- Examples of Tonicity adjuster/modifier are Glycerin, lactose, mannitol, dextrose, NaCl, sodium sulfate and sorbitol.
Importance of Isotonicity:
- An isotonic solution is one that exhibits the same effective osmotic pressure as blood serum.
- Isotonicity is important for parenteral preparation because when the solution is isotonic with blood, the possibility of product penetrating the RBC and causing haemolysis is reduced.
- For hypertonic solution crenation and for hypotonic solution haemolysis will occur.
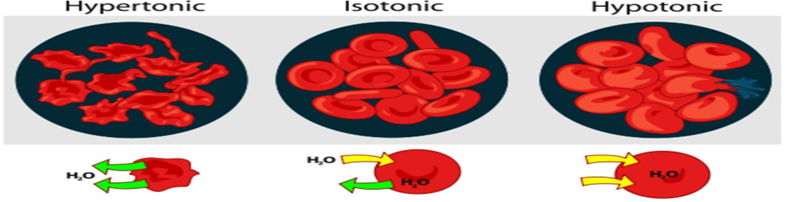
Why isotonicity is important:
- Prevents tissue damage
– Hypotonic solutions → cells swell and may burst (hemolysis).
– Hypertonic solutions → cells shrink (crenation)
– Isotonic solutions maintain normal cell shape and integrity. - Reduces pain and irritation
– Especially important for parenteral, ophthalmic, and nasal preparations.
– Isotonic formulations are more comfortable for patients. - Ensures patient safety
– Protects red blood cells and tissues during IV administration.
– Prevents serious adverse reactions. - Maintains normal physiological function
– Cells function best when osmotic balance is maintained.
– Supports proper diffusion and transport across membranes. - Improves drug effectiveness
– Proper isotonicity enhances drug absorption and therapeutic response.
Production procedure:
- Aseptic processing is the manufacture of sterile parenteral products by maintaining aseptic conditions throughout production — particularly during filling and sealing.
- It is used for heat- or radiation-sensitive drugs, biologicals and protein formulations.
- Heat sensitive or thermolabile drugs or Heat sensitive container and closure are used, terminal sterilization is not performed.
Selection & testing of raw materials
↓
Preparation of Water for Injection (WFI)
↓
Weighing and dispensing of API & excipients
↓
Bulk solution preparation
(API + WFI + excipients)
↓
pH adjustment & tonicity adjustment
↓
Pre-filtration (clarification)
↓
Sterilizing filtration (0.22 µm membrane)
↓
Cleaning, Sterilization of containers & closures
↓
Transfer to aseptic area (Grade A/B)
↓
Aseptic filling
↓
Stoppering / sealing
↓
Visual inspection
↓
Sterility, endotoxin & QC testing
↓
Labeling and packaging
1. Receipt, sampling & testing of materials
- APIs, excipients, solvents, containers and closures are identity-tested, checked for impurities, endotoxins and where applicable sterility.
- Only approved batches/lots are released to production.
2. WFI preparation & distribution
- Water for Injection (WFI) is produced (distillation / validated membrane system), stored/recirculated under conditions preventing microbial growth and periodically tested.
3. Weighing & dispensing
- Performed in controlled areas with restricted access; materials transferred through pass-boxes or air locks to avoid contamination.
4. Preparation of bulk solution (non-aseptic or aseptic compounding)
- API dissolved in WFI using non-sterilized or sterilized equipment; pH, tonicity and concentration adjusted; in-process sampling performed under controlled conditions.
5. Sterilizing filtration of bulk (0.22 µm membrane)
- Final bulk is passed through validated 0.22 µm sterilizing filters (integrity tested before and after use). Pre-filters used for clarification if required.
6. Washing and Sterilization of containers & closures
- Containers (vials, ampoules) and closures are cleaned and sterilized separately (dry-heat, steam or validated method), stored and transferred aseptically to filling area.
7. Aseptic filling & stoppering/sealing
- Filling performed under Grade A (laminar flow/isolator/RABS) with Grade B background. Minimal operator intervention, validated equipment, and closed systems where possible. Stoppering/crimping under aseptic conditions.
8 In-process controls & Visual inspection
- Fill volume, seal integrity, leaks and physical appearance. Samples taken for sterility, endotoxin and assay and Check for particulates.
9. Labelling, secondary packaging & release
- Performed in controlled area or after approved transfer. Final release based on QC results and batch documentation.
Production Facilities and Controls:
A parenteral manufacturing facility is designed to produce sterile, pyrogen-free medications for injection. The facility must minimize microbial, particulate, and endotoxin contamination.
Production Facilities (Design and Sections):
A standard parenteral production area is divided into five distinct/different sections to maintain environmental control:
- Clean-up Area.
- Preparation (Compounding) Area.
- Aseptic Area.
- Quarantine Area.
- Finishing & Packaging Area.
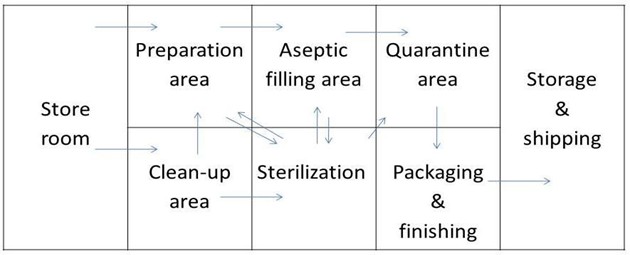
- Clean-up Area:
In a parenteral manufacturing facility, the Clean-up Area (also known as the Washing or Utensil Cleaning Area) is the designated zone for cleaning and decontaminating primary packaging components and manufacturing equipment.
- Core Functions:
- Decontamination: Removal of previous product residues, chemical contaminants, and environmental dust from equipment like mixing vessels, filter housings, and tubing.
- Waste Management: Safe disposal of broken glass rejected stoppers, and other process waste.
- Component Preparation: Initial washing of glass vials, ampoules, and rubber stoppers to remove particulates and “foreign matter” before they enter sterilization/ depyrogenation tunnels.
- Environmental Classification:
- Grade D (ISO 8): Unlike the aseptic filling zone, the clean-up area is generally a non-aseptic but highly controlled environment.
- Airflow: It is maintained under slightly negative pressure relative to adjacent corridors to prevent the spread of dust and detergents to cleaner zones.
- Surface Requirements: Walls and floors must be smooth, non-porous, and “coved” (rounded corners) to prevent water stagnation and facilitate easy disinfection.
- Specialized Equipment:
- Automated Washing Machines: Used for high-volume cleaning of vials and ampoules using pressurized Water for Injection (WFI).
- Ultrasonic Cleaners: Employs high-frequency sound waves to remove stubborn particles from intricate machine parts or needles.
- Drying Ovens: Hot air ovens used to dry equipment immediately after washing to prevent microbial growth in stagnant water.
- Cleaning Agents & Process:
- Water Quality: Initial cleaning often uses Purified Water, but the final rinse must be performed with Water for Injection (WFI) to ensure no ionic or microbial residues remain.
- Detergents: Standard agents include 1% sodium carbonate or specialized pharmaceutical-grade detergents (e.g., Teepol).
- Drying: Cleaned items are dried and immediately covered with lint-free bags or aluminum foil to prevent re-contamination before being moved to the sterilization area.
- Regulatory Requirements (cGMP):
- Sinks and Drains: While prohibited in Aseptic Areas, sinks are required here. However, they must be designed to prevent back-flow and be cleaned daily to avoid becoming a microbial reservoir.
- Storage: All cleaned equipment must be stored in a dry, dust-proof cabinet or covered area until needed for compounding.
- Logbooks: Every cleaning cycle must be documented in a Cleaning Record, including the agent used, batch number, and signature of the operator.
2. Preparation (compounding) area.
- Functional Roles:
- Compounding & Mixing: Dissolving active pharmaceutical ingredients (APIs) and excipients into the vehicle (e.g., water for injection) using specialized mixing tanks.
- Filtration: The primary stage where the bulk solution is passed through bacterial filters (typically 0.22 μm) to remove microorganisms and particulate matter before it reaches the filling area.
- Environmental Transition: Acting as a bridge between the non-sterile cleaning zones and the high-grade aseptic filling zone.
- Design and Engineering Requirements:
- HVAC & Airflow: Dedicated Heating, Ventilation, and Air Conditioning (HVAC) systems maintain positive pressure to ensure air flows out of cleaner areas, preventing ingress of contaminated air.
- HEPA Filtration: Air is continuously filtered through High-Efficiency Particulate Air (HEPA) filters, which remove at least 99.97% of airborne particles 0.3 μm or larger.
- Airlocks & Pass-Boxes: Personnel enter via gowning rooms (airlocks), and materials enter through interlocking pass-boxes to maintain the pressure differential.
- Cabinets and counters should be made of stainless steel and they are filled in such a way that no space is left for dirt to accumulate.
- Ceiling, walls and floor should be sealed and suitably painted to keep them thoroughly clean.
3. Aseptic area:
The aseptic area is the most critical zone in a parenteral production facility. It is a strictly controlled sterile environment designed to prevent microbial and particulate contamination during high-risk operations like aseptic connections, filling, and sealing.
- Classification and Air Quality:
- Grade A (ISO 5): The immediate area for high-risk operations (filling zone, open ampoules/vials). It must maintain a maximum of 3,520 particles (≥0.5 μm) per cubic meter both at rest and in operation.
- Grade B (ISO 5 at rest / ISO 7 in operation): The background environment surrounding Grade A zones, used for aseptic preparation and transport
- Design and Construction:
- The physical structure is engineered to be sterile and easily cleanable:
- Surface Integrity: Walls, floors, and ceilings must be smooth, waterproof, and non-shedding (e.g., epoxy-coated or stainless steel).
- Sealing: All joints must be sealed; coving is used at junctions to eliminate corners where dust can accumulate.
- Prohibitions: Drains and sinks are strictly forbidden in Grade A and B zones to prevent microbial growth from water sources.
- Airflow: Continuous Unidirectional (Laminar) Airflow at a velocity of 0.36–0.54 m/s ensures air moves in one direction to sweep away particles.
- Personnel and Gowning:
- Human operators are the primary source of contamination.
- PPE: Full sterile coveralls (a one-piece protective garment), hoods covering all hair, face masks, goggles, and sterile gloves (often double-gloving).
- Gowning Qualification: Personnel must undergo semi-annual gowning qualification tests using contact plates to ensure no microbes are shed during the process.
- Entry/Exit: Access is limited and strictly controlled via a series of airlocks (Black → Gray → White zones).
- Environmental Monitoring:
- Continuous monitoring is mandatory to ensure the area remains within sterile limits:
- Non-Viable Particulates: Continuous particle counting during operations to detect dust or fiber spikes.
- Viable Contaminants (Microbes): Measured using Settle Plates (passive air), Active Air Samplers (volumetric), and RODAC Plates for surface and finger-dab testing.
- Physical Parameters: Real-time monitoring of pressure differentials, temperature, and humidity.
- Cleaning and Disinfection:
- Sanitization: Use of sterile, low-lint wipes and a “clean-to-dirty” flow (Grade A first, then others).
- Disinfectant Rotation: Rotating two types of disinfectants plus a weekly sporicidal agent to prevent microbial resistance.
- Dwell Time: Surfaces must remain wet for the full “contact time” specified by the agent to ensure a complete kill.
4. Quarantine area:
In a parenteral production facility, the Quarantine Area is a designated, segregated zone used to temporarily hold materials or finished products until they are officially released for the next stage of processing or distribution.
- Isolation: Its primary role is to isolate products to prevent accidental use or distribution before Quality Control (QC) confirms they meet all safety and sterility standards.
- Checkpoint: It serves as a critical quality checkpoint where items are held pending results from tests like sterility, pyrogen, and bacterial endotoxin (BET).
- Risk Mitigation: By segregating unapproved stock, the area minimizes the risk of mix-ups and potential contamination of the supply chain
5. Finishing and packing area:
The finishing and packing area is the final stage of parenteral manufacturing where inspected and approved products are labeled and secondary-packaged for distribution.
- 100% Visual Inspection:
- Before labeling, every container must undergo mandatory inspection to ensure it is “essentially free” from visible particulates:
- Manual Inspection: Conducted against matte black and non-glare white backgrounds under intense, controlled light (2,000–3,750 lux). Operators must take mandatory rest breaks (typically every hour) to maintain visual acuity.
- Automated Inspection: Advanced facilities increasingly use automated systems, sometimes enhanced by Artificial Intelligence (AI), which must be validated to be equal to or better than human capability.
- Defect Detection: Containers are checked for extraneous particles (glass, fibers, metal), product precipitates, sealing/crimping defects, and glass cracks.
- Labeling Requirements:
- QR Codes: Mandatory for digital access to full product details, usage instructions, and safety warnings.
- High-Contrast Printing: Batch numbers and MRP must be printed with high-contrast inks for better readability
- Packaging Levels:
- Primary Packaging: The immediate container (ampoule, vial, pre-filled syringe) that must protect the sterile product from environmental and mechanical hazards.
- Secondary Packaging: Individual containers are placed into cardboard cartons or plastic partitioned boxes to prevent physical damage during transit.
- Tertiary Packaging: Shipper cases used for bulk transport. Standards are moving toward aggregation, where each carton is digitally linked to its shipper case for end-to-end traceability.
Controls:
Controls in parenteral (injectable) preparations are essential to ensure safety, sterility, and efficacy. These controls should be there in the entire manufacturing lifecycle, from raw materials to final packaging.
The following are the controls:
- Environmental and Facility Controls
- Personnel Controls
- In-Process Manufacturing Controls
- Finished Product Quality Control (QC) Tests
1. Environmental and Facility Controls
- Cleanroom Classification: Critical operations like filling occur in Grade A (Class 100) zones, where air contains no more than 100 particles per cubic foot.
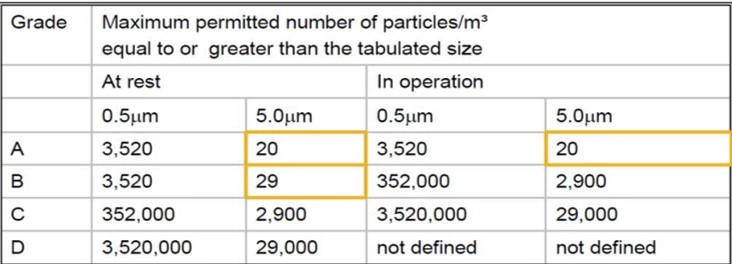
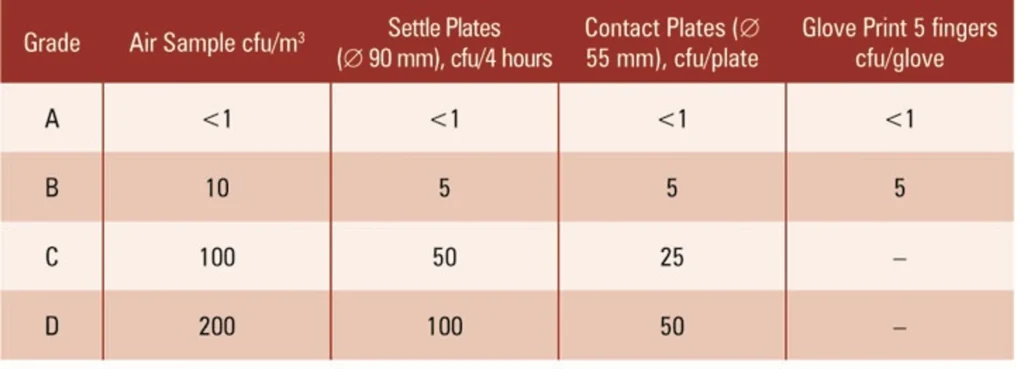
- Grade A: Below HEPA filter (inside filling machine)
- Grade B: Immediate after grade A, that is: outside the filling machine.

- Grade C: Usually compounding or preparation area.

- Grade D: Usually change rooms

- HEPA Filtration: High-Efficiency Particulate Air (HEPA) filters are used to remove at least 99.97% of airborne particles.
- Airflow Systems: Laminar Airflow (LAF) systems provide unidirectional air at specific velocities (approx. 0.45 m/s) to sweep contaminants away from the product.
- Differential Pressure: Aseptic areas are maintained at a higher positive pressure relative to adjacent rooms to prevent the ingress of outside air.
- Periodic Cleaning: Ceiling, walls, floor and every surface must be cleaned.
2. Personnel Controls:
- Humans are the primary source of contamination in sterile environments.
- Gowning: Personnel must wear specialized, non-shedding garments, including masks, hoods, gloves, and sterile suits.
- Training: Workers undergo rigorous training in aseptic techniques and hygiene.
- Monitoring: Regular sampling of gloves and gowns (e.g., using Rodac plates) ensures disinfection procedures are effective.
3. In-Process Manufacturing Controls:
- Monitoring occurs at every stage to ensure process stability and product quality.
- Environmental Monitoring: This involves continuously monitoring the temperature, humidity, and microbial count in the cleanroom environment where the product is manufactured and filled to ensure aseptic conditions are maintained.
- Bioburden Testing: Monitoring the microbial load of ingredients and the environment before sterilization.
- Water Quality: Continuous conductivity and temperature monitoring of Water for Injection (WFI) during distillation.
- Compounding: During compounding, there will be a QA guy to ensure the each and every step and instructions written in BMR is followed.
- pH and Conductivity Measurement: The pH and conductivity of the bulk solution are measured to ensure the vehicle is suitable for injection, compatible with the drug, and helps ensure product stability.
- Leakage Testing (Package Integrity): This test ensures the container seal is intact and free of capillary pores or micro-cracks that could allow microbial contamination. A common method is the “dye bath test”, where containers are immersed in a dye solution under vacuum and pressure, then inspected for dye ingress.
- Fill Volume/ weight uniformity: During the filling process, samples are periodically checked to confirm that the amount of liquid or powder in each container is accurate and uniform, meeting or slightly exceeding the labeled volume (e.g., within 100-110% of nominal volume).
- Visual inspection: Regular checks during filling is performed to ensure the product is free from foreign partials (Black, white, fiber, glass particles)and container-closure integrity.
- Aseptic Process Monitoring: Monitoring the aseptic behavior of the filling operator in aseptic area.
- Content Uniformity/Assay: Involves determining the active ingredient’s concentration in a random sample of containers to ensure it falls within the specified range (typically 85-115% of the average value).
4. Finished Product Quality Control (QC) Tests:
- Before release, every batch must pass several standardized tests.
- Sterility and Pyrogen Testing: While often considered final product quality control (FPQC), monitoring procedures like ongoing environmental controls help build confidence in sterility. Actual sterility and pyrogen (bacterial endotoxin) tests are critical evaluations of the final product, using methods like membrane filtration/direct inoculation for sterility and the Limulus Amebocyte Lysate (LAL) test for endotoxins.
- Particulate Matter Evaluation: Visual inspection or instrumental methods (e.g., light obscuration) to ensure products are free from visible and sub-visible particles.
- Leaker Testing: Used primarily for ampoules to detect seal defects, often by submerging them in a dye solution under vacuum.
- Uniformity of Content/Mass: Confirms that each dose contains the correct amount of active ingredients, especially for low-dose powders.
- pH and Isotonicity: Verifying the product’s pH (ideally 3.0–9.0) and tonicity to minimize tissue damage and pain upon injection.
Sterilization:
What is sterilization?
- Sterilization is the complete removal of microorganisms from an object or surfaces.
- Sterilization is obtained when microorganisms are subjected to antimicrobial agents for sufficient time and at optimum conditions.
Definition:
Sterilization is a process that completely eliminates, kills, or deactivates all forms of microbial life, including bacteria, viruses, fungi, and resistant spores, from a surface, object, fluid, or compound, making it safe for use or preventing infection.
Methods of sterilization:
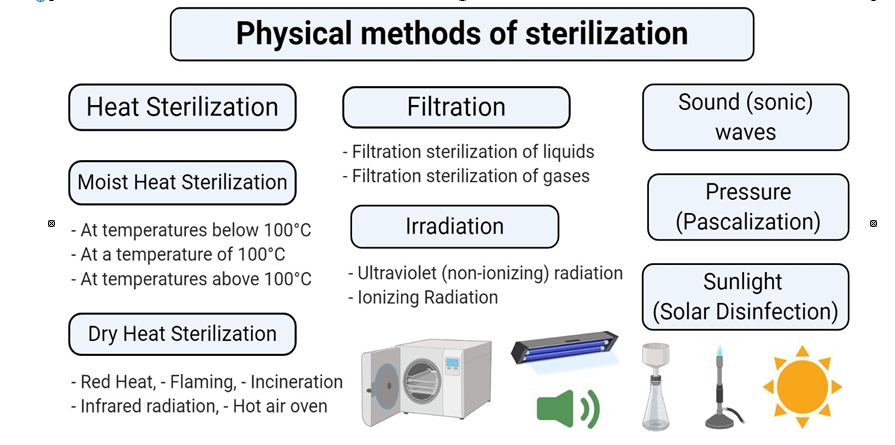
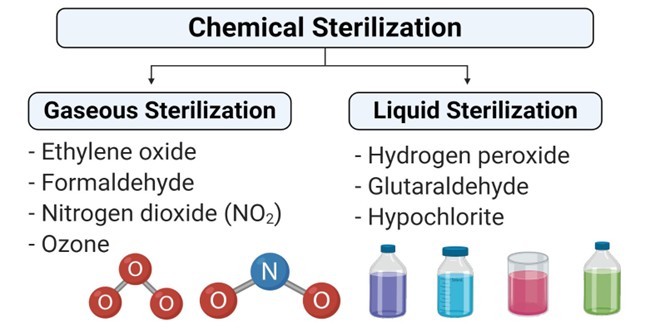
Physical methods of sterilization:
- Heat sterilization:
- Heat sterilization is the most effective and widely used method of sterilization, where the bactericidal activity results through the destruction of enzymes and other essential cell constituents.
- This method of sterilization is applicable to thermostable products. Still, it can be applied to both moisture-sensitive and moisture-resistant products, for which dry (160–180°C) and moist (121–134°C) heat sterilization procedures are respectively used.
- Moist Heat sterilization:
- Moist heat sterilization is one of the most effective methods of sterilization where the steam under pressure acts as a bactericidal agent.
- Moist heat sterilization usually involves the use of steam at temperatures in the range 121–134°C.
- High pressure increases the boiling point of water and thus helps achieve a higher temperature for sterilization.
- High pressure also facilitates the rapid penetration of heat into deeper parts of material and moisture present in the steam causes the coagulation of proteins causing an irreversible loss of function and activity of microbes.
Autoclave
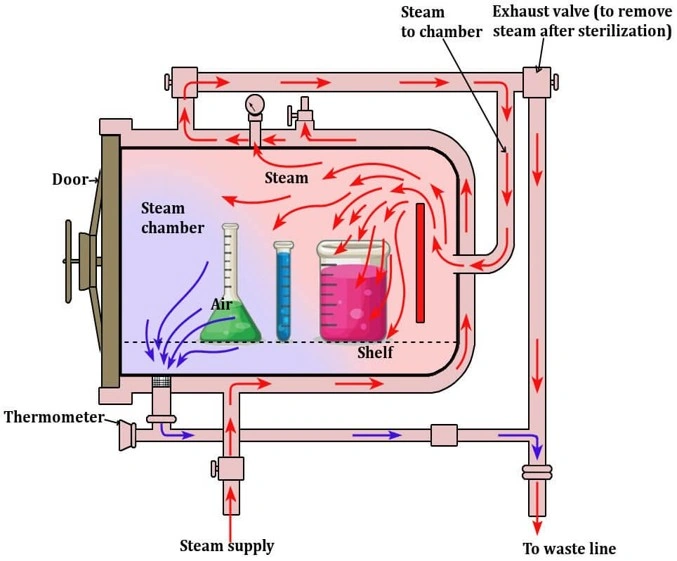

- The most commonly used standard temperature-time cycles for clinical porous specimens (e.g. surgical dressings) and bottled fluids are 134°C for 3 minutes and 121°C for 15 minutes, respectively.
- An autoclave is a device that works on the principle of moist heat sterilization through the generation of steam under pressure.
- In this method, the microorganisms are killed by coagulating their proteins, and this method is much more effective than dry heat sterilization where microbes are killed through oxidation.
- In the pharmaceutical and medical sectors, it is used in the sterilization of dressings, sheets, surgical and diagnostic equipment, containers, and aqueous injections, ophthalmic preparations, and irrigation fluids, in addition to the processing of soiled and contaminated items.
b. Dry heat sterilization:
- Dry sterilization is the process of removing microorganisms by applying moisture-free heat which is appropriate for moisture-sensitive substances.
- The dry heat sterilization process is based on the principle of conduction; that is the heat is absorbed by the outer surface of an item and then passed onward to the next layer. Ultimately, the entire item reaches the proper temperature needed to achieve sterilization.
- Dry, moisture-less heat destroys microorganisms by causing denaturation of proteins and also lyses the proteins in many organisms, causes oxidative free radical damage, causes dying of cells.
- Dry heat sterilization is used for the sterilization of materials which are difficult to sterilize by moist heat sterilization for several reasons.
- Substances like oil, powder, and related products cannot be sterilized by moist heat because moisture cannot penetrate into deeper parts of oily materials, and powders are destroyed by moisture.
- Similarly, laboratory equipment like Petri dishes and pipettes are challenging to sterilize by moist heat due to the penetration problem.
- The lethal effects of dry heat on microorganisms are primarily due to oxidative processes which are less effective when compared to the hydrolytic damage that results from exposure to steam in moist heat sterilization
Hot air oven

ii. Filtration:
- The process of filtration is unique among sterilization techniques in that it removes, rather than destroying microorganisms.
- Further, it is capable of preventing the passage of both viable and nonviable particles and can thus be used for both the clarification and sterilization of liquids and gases.
- The primary mechanisms involved in filtration are sieving, adsorption, and trapping within the matrix of the filter material.
- Filtration uses membranous filters that have tiny pores that let the Fluid pass through but prevent bigger particles such as bacteria from passing through the filter. Therefore, the smaller the pore, the more likely the filter is to stop more things from going through it.
0.2 micron Filters

iii. Irradiation:
- Irradiation is the process of exposing surfaces and objects to different kinds of radiation for sterilization.
- Mainly electromagnetic radiation is used for sterilization.
- The major target for these radiations is considered to be microbial DNA, where damage occurs as a result of ionization and free radical production (gamma-rays and electrons) or excitation (UV light).
Irradiation chamber

Chemical method of sterilization:
- Gaseous sterilization:
- Gaseous sterilization involves the process of exposing equipment or devices to different gases in a closed heated or pressurized chamber.
- Gaseous sterilization is a more effective technique as gases can pass through a tiny orifice and provide more effective results.
- Besides, gases are commonly used along with heat treatment which also facilitates the functioning of the gases.
- However, there is an issue of release of some toxic gases during the process which needs to be removed regularly from the system.
- The mechanism of action is different for different types of gases.


ii. Liquid sterilization:
- Liquid sterilization is the process of sterilization which involves the submerging of equipment in the liquid sterilant to kill all viable microorganisms and their spores.
- Although liquid sterilization is not as effective as gaseous sterilization, it is appropriate in conditions where a low level of contamination is present.
- Different liquid chemicals used for liquid sterilization.
- The surface decontamination is performed by the liquid sterilization method. Surfaces like ceiling, walls, floors, equipment surfaces and hand sanitization.
Quality Control Tests of Parenteral Products: –
Evaluation test for the parenteral:
The following are the evaluation test for the parenteral.
- Sterility test
- Clarity test
- Leakers test
- Pyrogen test
- Sterility test: It is a method carried out to detect confirm absence of any viable form of microbes in product. The method used for sterility tests are
a. Direct transfer method
b. Membrane filtration method.
a. Direct transfer method:
- Open each sample container and with draw the require amount of the sample.
- Inject one-half of sample in a test tube containing Fluid Thioglycolate Medium (FTM).
- Inject another half in the test tube containing Soyabean-casein digest Medium (SCDM).
- Volume of the medium must be sufficient to promote and expedite microbial growth.
- Adequate mixing between the sample inoculums and the culture medium must take place to maximize interaction and facilitate microbial growth.
- If the product to be tested contains any anti-microbial agent, using suitable reagent it should be neutralized before the test.
b. Membrane filtration method (MF):
- This method is employed in the following cases:
- Oil & oily preparations
- Alcoholic preparations
- For preparations miscible with or soluble in aqueous or oily solvents.
- The steps involved in MF sterility test method are
- The filter unit must be properly assembled and sterilized prior to use.
- The contents are transferred to the filter assembly under strict aseptic conditions.
- The membrane is removed aseptically.
- Membrane is cut in half.
- One half is place in suitable volume of Fluid Thioglycolate Medium (FTM) and another in an equal volume of Soyabean-casein digest Medium (SCDM).
2. Clarity test or 100% Visual Inspection:
- Manual Inspection: Conducted against matte black and non-glare white backgrounds under intense, controlled light (2,000–3,750 lux). Operators must take mandatory rest breaks (typically every hour) to maintain visual acuity.
- Automated Inspection: Advanced facilities increasingly use automated systems, sometimes enhanced by Artificial Intelligence (AI), which must be validated to be equal to or better than human capability.
- Defect Detection: Containers are checked for extraneous particles (glass, fibers, metal), product precipitates, sealing/crimping defects, and glass cracks.
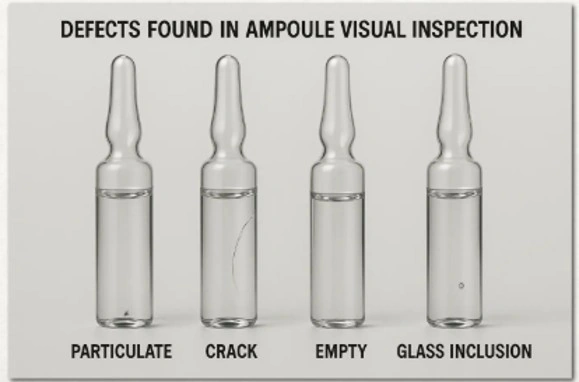
3. Leakers test:
- Leaker test for ampoules is intended to detect incompletely sealed ampoules so that they can be discarded in order to maintain sterile condition of the medicines. Open capillaries or cracks at the point of seal result in LEAKERS.
- The leaker test is performed by immersing the ampoules in a dye solution, such as 1% methylene blue, and applying at least 25 inches of vacuum for a minimum of 15 mins.
- Detection of leaker is prominent when ampoules are immersed in a bath of dye during autoclaving as this has advantage of accomplishing both leaker detection and sterilization in one operation.
- Another means of testing for leakers is a high frequency spark test system, which detect presence of pinholes in ampoules.
- Bottles and vials are not subjected to such a vacuum test because of the flexibility of the rubber closure
4. Pyrogen test:
A. The Pyrogen Test is a method to test the existence of pyrogens by using rabbits.
Test animals
- Use healthy mature rabbits each weighing not less than 1.5 kg which have not lost body mass when kept on a constant diet for not less than one week.
- Do not use the rabbits repeatedly in the same test unless as long a resting period as possible is taken.
- Animals should be excluded which have been used for a previous test that was decided as pyrogen positive.
- Record the rectal temperature four times at 2-hour intervals during 1 to 3 days prior to the test.
- House the animals individually during this period in an area free from disturbances likely to excite them, and take particular care to avoid disturbances on the day of the test.
- Keep the temperature in an area of performing test uniform between 20 0 C and 27 o C and preferably maintain constant humidity for at least 48 hours before the test.
Apparatus
- (1) Thermometer — Use a rectal thermometer or any other temperature – recording devices of equal sensitivity for which the time necessary for reading the rectal temperature is known.
- (2) Syringe and injection needle — Render the syringes and needles pyrogen-free by heating at 250 0 C for not less than 30 minutes.
Test procedures:
- Quantity of injection: unless otherwise specified, 10 mL of the sample per kg of body mass of the animal is used.
- Perform the test at an environmental temperature similar to that of the room wherein the animals were housed.
- The test animals are usually fixed in a suitable type of holder.
- Insert the thermometer or other temperature-recording device into the rectum of the test animal to a constant depth in the range of 60 to 90 mm, and read the temperature after a sufficient period of time.
- Withhold food from the test animals beginning several hours before the first temperature recording and until the test is completed.
- Determine the temperature of the test animals three times at 1hour intervals before the injection of the sample.
- When the second and third temperatures show little difference, the latter is taken as the “control temperature.“
- Do not use animals whose second and third temperatures are not in accord or exceed 39.8 0 C even if these two values are similar.
- Warm the sample to 37 o c before injection, and administer intravenously through an ear vein within 15 minutes after the third temperature recording.
- Hypotonic solution other than Water for Injection may be made isotonic by the addition of pyrogen-free sodium chloride before the test.
- Read the temperatures three times at I-hour intervals after injection.
- The difference between the control temperature and the highest temperature is taken to be the rise in body temperature.
Interpretation of results:
- The test is carried out on a group of three rabbits.
- The test shall be considered positive, if two or more of the five rabbits show an individual temperature rise of 0.6 0 C or more.
- When the pyrogen test is positive, the sample is considered to be rejected.
- If two or three rabbits show an individual rise of 0.6 0 C or more above the respective control temperature, the test shall be considered as positive.
- If only one animal shows a temperature rise of 0.6 0 C or more, or if the sum of the temperature rises of the three animals exceeds 1.4 0 C, repeat the test on a group of five other rabbits.
B. The Pyrogen Test is a method to test the existence of pyrogens by using Limulus Amoebocyte Lysate.
Bacterial Endotoxin Test (BET) or Limulus Amoebocyte Lysate Test (LAL Test):-
- The bacterial endotoxin test (BET) is a test to detect or quantify endotoxins from gram negative bacteria using Amoebocyte lysate from the horse shoe crab.
- The endotoxins of gram-negative bacteria forms a firm gel within 60 mins in the presence of lysate of amebocytes of limulus polyphemus of horseshoe crab, when incubated at 37◦c.
- Hence, the test is only effective with gram-negative bacteria, which constitute the majority and the most potent of the pyrogens.
- The addition of a solution containing endotoxins to a solution of a lysate produces turbidity, precipitation or gelation of the mixture.
Dry powder preparation by Lyophilization:
Parenterals are drugs administered by injection. Many of these drugs chemically degrade if kept in water (by hydrolysis).
To solve this, manufacturers remove the water to create a stable dry powder or “cake.” This powder is stored in a vial and must be reconstituted by mixing with a sterile diluent like WFI or saline immediately before injection.
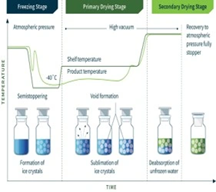
The Manufacturing Process: Lyophilization
The machine used for lyophilization is Lyophilizer (Freeze Dryer). The process relies on sublimation, where ice turns directly into vapor without becoming liquid.

The process consists of five main stages:
- Preparation & Filling.
- Freezing (Solidification).
- Primary Drying (Sublimation).
- Secondary Drying (Desorption).
- Sealing.
1. Preparation & Filling:
- The drug is dissolved in a solvent (usually Water for Injection) and sterilized by passing it through a 0.22-micron filter.
- The sterile solution is filled into vials.
- Crucial Step: Special slotted stoppers are placed partially into the vial neck to allow water vapor to escape later.
2. Freezing (Solidification):
- The vials are loaded onto cooled shelves in the lyophilizer.
- The temperature is lowered (often -40°C to -50°C) until the solution freezes completely.
- Academic Note: The product must be cooled below its “triple point” or “eutectic temperature” to ensure complete freezing.
3. Primary Drying (Sublimation):
- A vacuum is applied to the chamber to lower the pressure.
- The shelves are gently heated.
- Because of the low pressure, the ice sublimes (turns to gas) and moves to a condenser where it turns back to ice.
- This step removes the majority of the water (unbound water).
4. Secondary Drying (Desorption):
- The shelf temperature is raised further (sometimes to ambient or higher) while maintaining a high vacuum.
- This removes “bound water” (water molecules chemically attached to the drug).
- This ensures the final moisture content is very low (often <1-3%) for stability.
5. Sealing:
- Hydraulic plates in the machine push the stoppers completely down while still under vacuum (or inert gas atmosphere), sealing the sterile environment inside the vial.
Advantages (Why use Lyophilization?)
- Stability: It prevents hydrolysis, allowing heat-sensitive drugs to be stored at room temperature for years.
- Rapid Reconstitution: The resulting “cake” is porous and dissolves almost instantly when diluent (sterile WFI or saline) is added.
- Precision: It maintains the exact dose filled into the vial, unlike powder filling which can vary.
- Sterility: The process is a closed system, reducing contamination risk.
Disadvantages
- Cost: Lyophilizers are expensive and consume high amounts of energy.
- Time: The drying cycle is slow, often taking 24 to 48 hours or longer.
- Handling: The final product requires the nurse or doctor to manually mix it before use.
Common Examples
- Antibiotics: Penicillin, Cephalosporins, Doxycycline.
- Biologics: Vaccines, Monoclonal Antibodies, Insulin.
Define ophthalmic preparation with examples:
Ophthalmic preparations are specialized, sterile dosage forms designed to be instilled onto the external surface of the eye (topical), administered inside the eye (intraocular), or placed adjacent to it (periocular). As the eye is a highly sensitive organ with poor natural defense barriers, these preparations must meet strict pharmaceutical standards, most notably sterility and isotonicity (matching the osmotic pressure of tears).
Classification and Examples
Ophthalmic preparations are generally classified by their physical state and mode of delivery:
1. Ophthalmic Solutions (Eye Drops)
- These are the most common forms. They are sterile, clear liquids free from foreign particles.
- Definition: The active drug is completely dissolved in an aqueous (water-based) or oily vehicle.
- Examples:
- Antibiotics: Moxifloxacin Eye Drops (for bacterial infections).
- Glaucoma treatment: Timolol Maleate Eye Drops (to lower intraocular pressure).
- Diagnostics: Sodium Fluorescein (dye used to detect corneal damage).
Classification and Examples
Ophthalmic preparations are generally classified by their physical state and mode of delivery:
1. Ophthalmic Solutions (Eye Drops)
These are the most common forms. They are sterile, clear liquids free from foreign particles.
- Definition: The active drug is completely dissolved in an aqueous (water-based) or oily vehicle.
- Examples:
- Antibiotics: Moxifloxacin Eye Drops (for bacterial infections).
- Glaucoma treatment: Timolol Maleate Eye Drops (to lower intraocular pressure).
- Diagnostics: Sodium Fluorescein (dye used to detect corneal damage).
2. Ophthalmic Suspensions
Used when the drug is not soluble in water or to improve stability.
- Definition: A sterile liquid containing finely divided solid particles dispersed in a vehicle. The particles must be ultra-fine (<10 microns) to prevent gritty irritation in the eye.
- Examples:
- Corticosteroids: Prednisolone Acetate Ophthalmic Suspension (anti-inflammatory).
- Combination drugs: Tobramycin and Dexamethasone suspension.
3. Ophthalmic Ointments
These are semi-solid preparations used for prolonged contact with the eye.
- Definition: The drug is incorporated into a sterile base (usually a mixture of mineral oil and white petrolatum) that melts at body temperature. They are often used at night because they can cause temporary blurred vision.
- Examples:
- Antibiotic: Chloramphenicol Eye Ointment.
- Lubricant: Simple sterile petrolatum ointment for dry eyes.
4. Ophthalmic Gels (Viscous Solutions)
- Definition: These use polymers (like carbomer or methylcellulose) to increase viscosity. This keeps the drug in contact with the eye longer than standard drops without the blurriness of ointments.
- Examples:
- Artificial Tears: Carboxymethylcellulose (CMC) Gel.
- Treatment: Ganciclovir Ophthalmic Gel (antiviral).
5. Ocular Inserts
A newer technology designed for controlled, continuous release.
- Definition: Sterile, thin, solid or semi-solid wafers placed in the cul-de-sac (under the eyelid). They dissolve or release drug over a period of days.
- Example: Ocusert Pilo-20 (delivers pilocarpine for glaucoma over 7 days).